Alpha-Mannosidosis: Therapeutic Strategies

 ,
, 

Abstract
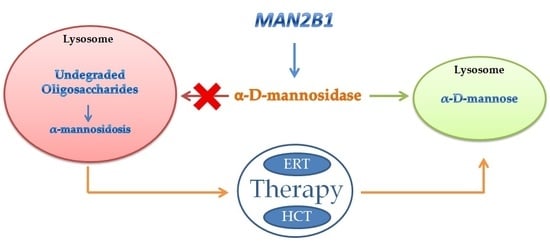
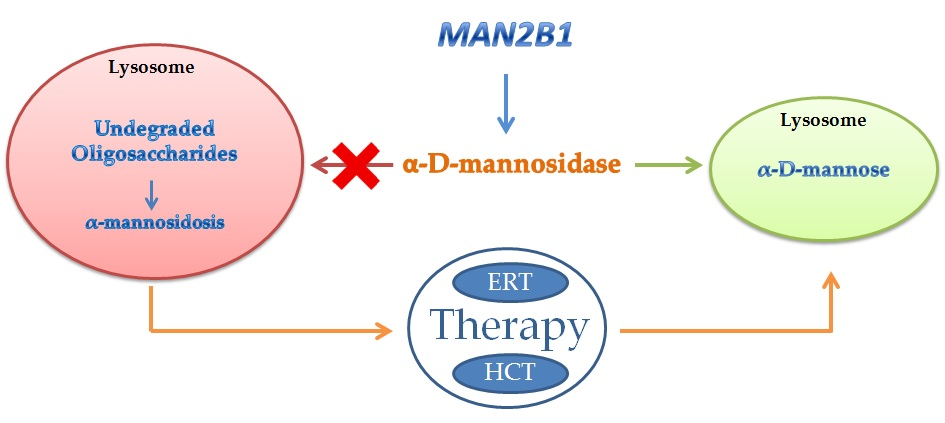{kind=link}
1. Introduction
2. Inherited Deficiency of Alpha-Mannosidase (Alpha-Mannosidosis)
3. Genetics of Alpha-Mannosidase
4. Therapy of Alpha-Mannosidosis
Bone Marrow Transplantation
5. Enzyme Replacement Therapy
Conflicts of Interest
References
- Winchester, B. Lysosomal metabolism of glycoproteins. Glycobiology 2005, 15, 1R–15R. [Google Scholar] [CrossRef] [PubMed]
- Persichetti, E.; Klein, K.; Paciotti, S.; Lecointe, K.; Balducci, C.; Franken, S.; Duvet, S.; Matzner, U.; Roberti, R.; Hartmann, D.; et al. Lysosomal di-N-acetylchitobiase-deficient mouse tissues accumulate Man2GlcNac2 and Man3GlcNac2. Biochim. Biophys. Acta 2012, 1822, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug. Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Malcolm, S.; Pemble, S.; Winchester, B. Purification and comparison of the structures of human liver acidic alpha-d-mannosidase A and B. Biochem. J. 1986, 233, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Beccari, T.; Stinchi, S.; Orlacchio, A. Lysosomal alpha-mannosidase. Biosci. Rep. 1999, 19, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Beccari, T.; Appolloni, M.G.; Costanzi, E.; Stinchi, S.; Stirling, J.L.; Della Fazia, M.A.; Servillo, G.; Viola, M.P.; Orlacchio, A. Lysosomal alpha-mannosidase of mouse tissues: Characteristic of the isoenzymes, and cloning and expression of a full-length cDNA. Biochem. J. 1997, 327, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Ockerman, P.A. A generalized storage disorder resembling Hurler’s syndrome. Lancet 1967, 2, 239–241. [Google Scholar] [CrossRef]
- Hocking, J.D.; Jolly, R.D.; Batt, R.D. Deficiency of alpha-mannosidase in Angus cattle. An inherited lysosomal storage disease. Biochem. J. 1972, 128, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Burditt, L.J.; Chotai, K.; Hirani, S.; Nugent, P.G.; Winchester, B.G.; Blackmore, W.F. Biochemical studies on a case of feline mannosidosis. Biochem. J. 1980, 189, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Crawley, A.C.; Jones, M.Z.; Bonning, L.E.; Finnie, J.W.; Hopwood, J.J. α-mannosidosis in the guinea pig: A new animal model for lysosomal storage disorders. Pediatr. Res. 1999, 46, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Huxtable, C.R.; Dorling, P.R.; Walkley, S.U. Onset and regression of neuroaxonal lesions in sheep with mannosidosis induce experimentally with swainsonine. Acta Neuropathol. 1982, 58, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Stinchi, S.; Lüllmann-Rauch, R.; Hartmann, D.; Coenen, R.; Beccari, T.; Orlacchio, A.; von Figura, K.; Saftig, P. Targeted disruption of the lysosomal α-mannosidase gene results in mice resembling a mild form of human α-mannosidosis. Hum. Mol. Genet. 1999, 8, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Poupetová, H.; Ledvinová, J.; Berná, L.; Dvoráková, L.; Kozich, V.; Elleder, M. The birth prevalence of lysosmal storage disorders in the Czech republic: Comparison with data in different populations. J. Inherit. Metab. Dis. 2010, 33, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Malm, D.; Nilssen, O. Alpha-mannosidosis. Orphan. J. Rare Dis. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Olsen, K.J.; Wraith, J.E.; Zeman, J.; Michalski, J.C.; Saftig, P.; Fogh, J.; Malm, D. Natural history of alpha-mannosidosis a longitudinal study. Orphan. J. Rare Dis. 2013, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Riise, H.M.; Hansen, G.M.; Malm, D.; Tranebjaerg, L.; Tollersrud, O.K.; Nilssen, O. Spectrum of mutations in alpha-mannosidosis. Am. J. Hum. Genet. 1999, 64, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J.; Lewandowski, R.C., Jr.; Sanfilippo, S.J.; Tsai, M.Y.; Foni, I.; Bruhl, H.H. Clinical manifestation of alpha-mannosidosis: A longitudinal study. Am. J. Med. 1976, 61, 841–848. [Google Scholar] [CrossRef]
- Ara, J.R.; Mayayo, E.; Marzo, M.E.; Guelbenzu, S.; Chabás, A.; Pina, M.A.; Calderón, C. Neurological impairment in α-mannosidosis: A longitudinal clinical and MRI study of a brother and sister. Childs Nerv. Syst. 1999, 15, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Noll, R.B.; Netzloff, M.L.; Kulkarni, R. Long-term follow-up of biochemical and cognitive funtioning in patients with alpha-mannosidosis. Arch. Neurol. 1989, 46, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Chester, M.A.; Lundblad, A.; Ockerman, P.A.; Autio, S. Mannosidosis. In Genetic Errors of Glycoprotein Metabolism; Durand, P., O’Brien, J.F., Eds.; Edi-Ermes: Milan, Italy, 1982; pp. 89–122. ISBN 8870510050, 9788870510058. [Google Scholar]
- Autio, S.; Louhimo, T.; Helenius, M. The clinical course of mannosidosis. Ann. Clin. Res. 1982, 14, 93–97. [Google Scholar] [PubMed]
- Swiedler, S.J.; Beck, M.; Bajbouj, M.; Giugliani, R.; Schwartz, I.; Harmatz, P.; Wraith, J.E.; Roberts, J.; Ketteridge, D.; Hopwood, J.J.; et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey with Mucopolisaccharidosis VI (Maroteaux-Lamy syndrome). Am. J. Med. Genet. A 2005, 134A, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Champion, M.J.; Shows, T.B. Mannosidosis: Assignment of the lysosomal alpha-mannosidase B gene to chromosome 19 in man. Proc. Natl. Acad. Sci. USA 1977, 74, 2968–2972. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, Y.; Hayes, H.; Uchida, T.; Michihiro, M.C.; Okada, Y. Regional assignment of five genes on human chromosome 19. Am. J. Hum. Genet. 1998, 63, 1015–1024. [Google Scholar] [CrossRef]
- Nebes, B.L.; Schmidt, M.C. Human lysosomal alpha-mannosidase: Isolation and nucleotide sequence of the full-length cDNA. Biochem. Biophys. Res. Commun. 1994, 200, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.F.; Lal, A.L.; Moremen, K.W. Cloning, expression, purification, and characterization of the human broad specificity lysosomal acid α-mannosidase. J. Biol. Chem. 1996, 8, 28348–28358. [Google Scholar] [CrossRef]
- Nilssen, O.; Berg, T.; Riise, H.M.F.; Ramachandran, U.; Evjen, G.; Hansen, G.M.; Malm, D.; Tranebjaerg, L.; Tollersrud, O.K. α-mannosidosis: Functional cloning of the lysosomal α-mannosidase cDNA and identification of a mutation in two affected siblings. Hum. Mol. Genet. 1997, 6, 717–726. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kozak, M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 1987, 196, 947–950. [Google Scholar] [CrossRef]
- Riise, H.M.; Berg, T.; Nilssen, O.; Romeo, G.; Tollersrud, O.K.; Ceccherini, I. Genomic structure of the human lysosomal α-mannosidase gene (MANB). Genomics 1997, 42, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, N.; Gotoda, Y.; Saito, S.; Kawai, H. Characterization of the human MANB gene encoding lysosomal α-d-mannosidase. Gene 1997, 198, 351–357. [Google Scholar] [CrossRef]
- Tollersrud, O.K.; Berg, T.; Healy, P.; Evjen, G.; Ramachandran, U.; Nilssen, O. Purification of bovine alpha-mannosidase, characterization of its gene and determination of two mutations that cause alpha-mannosidosis. Eur. J. Biochem. 1997, 246, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Tollersrud, O.K.; Walkley, S.U.; Siegel, D.; Nilssen, O. Purification of feline lysosomal alpha-mannosidase, determination of its cDNA sequence and identification of a mutation causing alpha-mannosidosis in Persian cats. Biochem. J. 1997, 328, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Hopwood, J.J. alpha-Mannosidosis in the guinea pig: Cloning of the lysosomal α-mannosidase cDNA and identification of a missense mutation causing α-mannosidosis. Biochim. Bipphys. Acta 2002, 1586, 169–176. [Google Scholar] [CrossRef]
- Gotoda, Y.; Wakamatsu, N.; Kawai, H.; Nishida, Y.; Matsumoto, T. Missense and nonsense mutations in the lysosomal alpha-mannosidase gene (MANB) in severe and mild forms of alpha-mannosidosis. Am. J. Hum. Genet. 1998, 63, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Beccari, T.; Bibi, L.; Ricci, R.; Antuzzi, D.; Burgalossi, A.; Costanzi, E.; Orlacchio, A. Two novel mutations in the gene for human α-mannosidase that cause α-mannosidosis. J. Inherit. Metab. Dis. 2003, 26, 819–820. [Google Scholar] [CrossRef] [PubMed]
- Urushihara, M.; Kagami, S.; Yasutomo, K.; Ito, M.; Kondo, S.; Kitamura, A.; Malm, D.; Klenow, H.; Nilssen, O.; Kuroda, Y. Sisters with α-mannosidosis and systemic lupus erythematosus. Eur. J. Pediatr. 2004, 163, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Sbaragli, M.; Bibi, L.; Pittis, M.G.; Balducci, C.; Heikinheimo, P.; Ricci, R.; Antuzzi, D.; Parini, R.; Spaccini, L.; Bembi, B.; et al. Identification and characterization of five novel MAN2B1 mutations in Italian patients with alpha-mannosidosis. Hum. Mutat. 2005, 25, 320. [Google Scholar] [CrossRef] [PubMed]
- Castelnovo, G.; Levade, T.; Riise Stensland, H.M.; Nonnon, M.J.; Berges, M.A.; Tollersrud, O.K.; Labauge, P. Adult leukoencephalopathy caused by alpha-mannosidosis deficiency. Rev. Neurol. 2007, 163, 359–361. [Google Scholar] [CrossRef]
- Lyons, M.J.; Wood, T.; Espinoza, L.; Stensland, H.M.; Holden, K.R. Early onset alpha-mannosidosis with slow progression in three Hispanic males. Dev. Med. Child. Neurol. 2007, 49, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Pittis, M.G.; Montalvo, A.L.E.; Heikinheimo, P.; Sbaragli, M.; Balducci, C.; Persichetti, E.; Van Maldergem, L.; Filocamo, M.; Bembi, B.; Beccari, T. Funtional characterization of four novel MAN2B1 mutations causing juvenile onset alpha-mannosidosis. Clin. Chim. Acta 2007, 375, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Kuokkanen, E.; Riise Stensland, H.M.; Smith, W.; Kjeldsen Buvang, E.; Van Nguyen, L.; Nilssen, O.; Heikinheimo, P. Molecular and cellular characterization of novel α-mannosidosis mutations. Hum. Mol. Genet. 2011, 20, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Riise Stensland, H.M.; Klenow, H.B.; Van Nguyen, L.; Hansen, G.M.; Malm, D.; Nilssen, O. Identification of 83 novel alpha-mannosidosis-associated sequence variants: Functional analysis of MAN2B1 missense mutations. Hum. Mutat. 2012, 33, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pan, J.; Guo, Y.; Guo, C.; Jiang, W.; Li, R.; Tang, J.; Ai, Y. Molecular diagnosis of a chinese pedigree with alpha-mannosidosis and identification of a novel missense mutation. J. Pediatr. Endocrinol. Metab. 2014, 27, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Riise Stensland, H.M.; Frantzen, G.; Kuokkanen, E.; Buvang, E.K.; Klenow, H.B.; Heikinheimo, P.; Malm, D.; Nilssen, Ø. amamutdb.no: A relational database for MAN2B1 allelic variants that compiles genotypes, clinical phenotypes, and biochemical and structural data of mutant MAN2B1 in α-mannosidosis. Hum. Mutat. 2015, 36, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Krivit, W.; Lockman, L.; Jambaqué, I.; Peters, C.; Cowan, M.; Harris, R.; Blanche, S.; Bordigoni, P.; Loes, D.; et al. Long-term effect of bone-marrow transplantation for chilhood-onset cerebral X-linked adrenoleukodystrohy. Lancet 2000, 356, 713–718. [Google Scholar] [CrossRef]
- Walkley, S.U.; Thrall, M.A.; Dobrenis, K.; Huang, M.; March, P.A.; Siegel, D.A.; Wurzelmann, S. Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. Proc. Natl. Acad. Sci. USA 1994, 91, 2970–2974. [Google Scholar] [CrossRef] [PubMed]
- Will, A.; Cooper, A.; Hatton, C.; Sardharwalla, I.B.; Evans, D.I.; Stevens, R.F. Bone marrow transplantation in the treatment of alpha-mannosidosis. Arch. Dis. Child. 1987, 62, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.A.; Grange, D.K.; Goulding, P.; Daines, M.; Luisiri, A.; Kotagal, S. Bone marrow transplantation for the treatment of alpha-mannosidosis. J. Pediatr. 1998, 133, 282–285. [Google Scholar] [CrossRef]
- Grewal, S.S.; Shapiro, E.G.; Krivit, W.; Charnas, L.; Lockman, L.A.; Delaney, K.A.; Davies, S.M.; Wenger, D.A.; Rimell, F.L.; Abel, S.; et al. Effective treatment of α-mannosidosis by allogenic hematopoietic stem cell transplantation. J. Pediatr. 2004, 144, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.H.; Schuster, F.; Peters, C.; Schulze, S.; Pontz, B.F.; Muntau, A.C.; Röschinger, W.; Stachel, D.K.; Enders, A.; Haas, R.J.; et al. T-cell-depleted peripheral blood stem cell transplantatio for α-mannosidosis. Bone Marrow Transpl. 2003, 32, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.A.; Chakrapani, A.; Wraith, J.E. The effects of early and late bone marrow transplantation in siblings with alpha-mannosidosis. Is early haematopoietic cell transplantation the preferred treatment option? J. Inherit. Metab. Dis. 2010, 33, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Yesilipek, A.M.; Akcan, M.; Karuso, G.; Uygun, V.; Kupesiz, A.; Hazar, V. Successful unrelated bone marrow transplantation in two siblings with alpha-mannosidosis. Pediatr. Transpl. 2012, 16, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Mynarek, M.; Tolar, J.; Albert, M.H.; Escolar, M.L.; Boelens, J.J.; Cowan, M.J.; Finnegan, N.; Glomstein, A.; Jacobsohn, D.A.; Kühl, J.S.; et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: An analysis of 17 patients. Bone Marrow Transpl. 2012, 47, 352–359. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.A.; Eastlund, T.; Peters, C.; Neglia, J.P.; Defor, T.; Ramsay, N.K.; Scott Baker, K. Autoimmune haemolytic anaemia complicating haematopoitic cell transplantation in paediatric patients: high incidence and significant mortality in unrelated donor transplants for non-malignant diseases. Br. J. Haematol. 2004, 127, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Panoskaltsis-Mortari, A.; Haddad, I.Y.; Blazar, B.R.; Orchard, P.J.; Cornfield, D.N.; Grewal, S.S.; Peters, C.; Regelmann, W.E.; Milla, C.E.; et al. Inflammatory cytokines and the development of pulmonary complications after allogeneic hematopoitic cell transplantation with inherited metabolic storage disorders. Biol. Blood Marrow Transpl. 2006. 12, 430–437. [CrossRef]
- Staba, S.L.; Escolar, M.L.; Poe, M.; Kim, Y.; Martin, P.L.; Szabolcs, P.; Allison-Thacker, J.; Wood, S.; Wenger, D.A.; Rubinstein, P.; et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N. Engl. J. Med. 2004, 350, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.; Muro, S. Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives. Adv. Drug. Deliv. Rev. 2017, 118, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Roces, D.P.; Lüllmann-Rauch, R.; Peng, J.; Balducci, C.; Andersson, C.; Tollersrud, O.; Fogh, J.; Orlacchio, A.; Beccari, T.; Saftig, P.; et al. Efficacy of enzyme replacement therapy in α-mannosidosis mice: A preclinical animal study. Hum. Mol. Genet. 2004, 13, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Blanz, J.; Stroobants, S.; Lüllmann-Rauch, R.; Morelle, W.; Lüdemann, M.; D’Hooge, R.; Reuterwall, H.; Michalski, J.C.; Fogh, J.; Andersson, C.; Saftig, P. Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in α-mannosidosis mice. Hum. Mol. Genet. 2008, 17, 3437–3445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Urayama, A.; Grubb, J.H.; Banks, W.A.; Sly, W.S. Epinephrine enhances lysosomal enzyme delivery across the blood brain barrier by up-regulation of the mannose-phospahte receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Crawley, A.C.; King, B.; Berg, T. Enzyme replacement therapy in α-mannosidosis guinea pigs. Mol. Genet. Metab. 2006, 89, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Courenay, A.; Troendle, F.J.; Stallings-Mann, M.L.; Dickey, C.A.; De Lucia, M.W.; Dickson, D.W.; Eckman, C.B. Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. FASEB J. 2005, 19, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Matzner, U.; Lullmann-Rauch, R.; Stroobants, S.; Andersson, C.; Weigelt, C.; Eistrup, C.; Fogh, J.; D’Hooge, R.; Gieselmann, V. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leucodystrophy. Mol. Ther. 2009, 17, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Baldo, G.; Giugliani, R.; Matte, U. Lysosomal enzymes may cross the blood-brain-barrier by pinocytosis: implication for enzyme replacement therapy. Med. Hypotheses 2014, 82, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Vogler, C.; Levy, B.; Grubb, J.H.; Galvin, N.; Tan, Y.; Kakkis, E.; Pavloff, N.; Sly, W.S. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA 2005, 102, 14777–14782. [Google Scholar] [CrossRef] [PubMed]
- Damme, M.; Stroobants, S.; Lüdemann, M.; Rothaug, M.; Lüllmann-Rauch, R.; Beck, H.C.; Ericsson, A.; Andersson, C.; Fogh, J.; D’Hooge, R.; et al. Chronic enzyme replacement therapy ameliorates neuropathology in alpha-mannosidosis mice. Ann. Clin. Transl. Neurol. 2015, 2, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Stroobants, S.; Damme, M.; Van der Jeugd, A.; Vermaercke, B.; Andersson, C.; Fogh, J.; Saftig, P.; Blanz, J.; D’Hooge, R. Long-term replacement therapy improves neurocognitive functioning and hippocampal synaptic plasticity in immune-tolerant alpha-mannosidase mice. Neurobiol. Dis. 2017, 106, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Borgwardt, L.; Dali, C.I.; Fogh, J.; Månsson, J.E.; Olsen, K.J.; Beck, H.C.; Nielsen, K.G.; Nielsen, L.H.; Olsen, S.O.; Riise Stensland, H.M.; et al. Enzyme replacement therapy for alpha-mannosidosis: 12 months follow-up of a single centre, randomised, multiple dose study. J. Inherit. Metab. Dis. 2013, 36, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Borgwardt, L.; Lund, A.M.; Amraoui, Y.; Andersen, O.; De Meirleir, L.; Dolhem, P.; Campos, M.G.; Guffon, N.; Héron, B.; Laroche, C.; et al. Long-term enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase) slows disease progression in adult patients suffering from alpha-mannosidosis. Mol. Genet. Metab. 2017, 120, s30. [Google Scholar] [CrossRef]
- Borgwardt, L.; Lund, A.M.; Amraoui, Y.; Andersen, O.; De Meirleir, L.; Dolhem, P.; Campos, M.G.; Guffon, N.; Héron, B.; Jameson, E.; et al. Improvement in fine and gross motor proficiency after long-term enzyme replacement therapy with velmanase alfa (human recombinant alpha mannosidase) in alpha-mannosidosis patients. Mol. Genet. Metab. 2017, 120, s29. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccarini, M.R.; Codini, M.; Conte, C.; Patria, F.; Cataldi, S.; Bertelli, M.; Albi, E.; Beccari, T. Alpha-Mannosidosis: Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 1500. https://doi.org/10.3390/ijms19051500
Ceccarini MR, Codini M, Conte C, Patria F, Cataldi S, Bertelli M, Albi E, Beccari T. Alpha-Mannosidosis: Therapeutic Strategies. International Journal of Molecular Sciences. 2018; 19(5):1500. https://doi.org/10.3390/ijms19051500
Chicago/Turabian StyleCeccarini, Maria Rachele, Michela Codini, Carmela Conte, Federica Patria, Samuela Cataldi, Matteo Bertelli, Elisabetta Albi, and Tommaso Beccari. 2018. "Alpha-Mannosidosis: Therapeutic Strategies" International Journal of Molecular Sciences 19, no. 5: 1500. https://doi.org/10.3390/ijms19051500
APA StyleCeccarini, M. R., Codini, M., Conte, C., Patria, F., Cataldi, S., Bertelli, M., Albi, E., & Beccari, T. (2018). Alpha-Mannosidosis: Therapeutic Strategies. International Journal of Molecular Sciences, 19(5), 1500. https://doi.org/10.3390/ijms19051500