Involvement of MAFB and MAFF in Retinoid-Mediated Suppression of Hepatocellular Carcinoma Invasion


Abstract
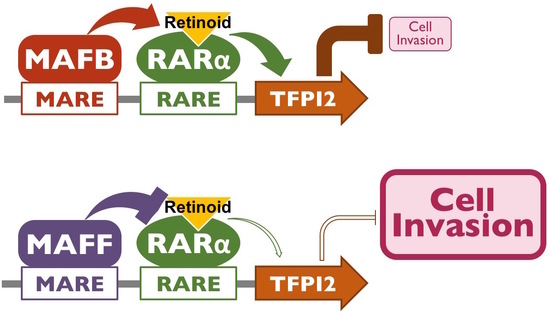
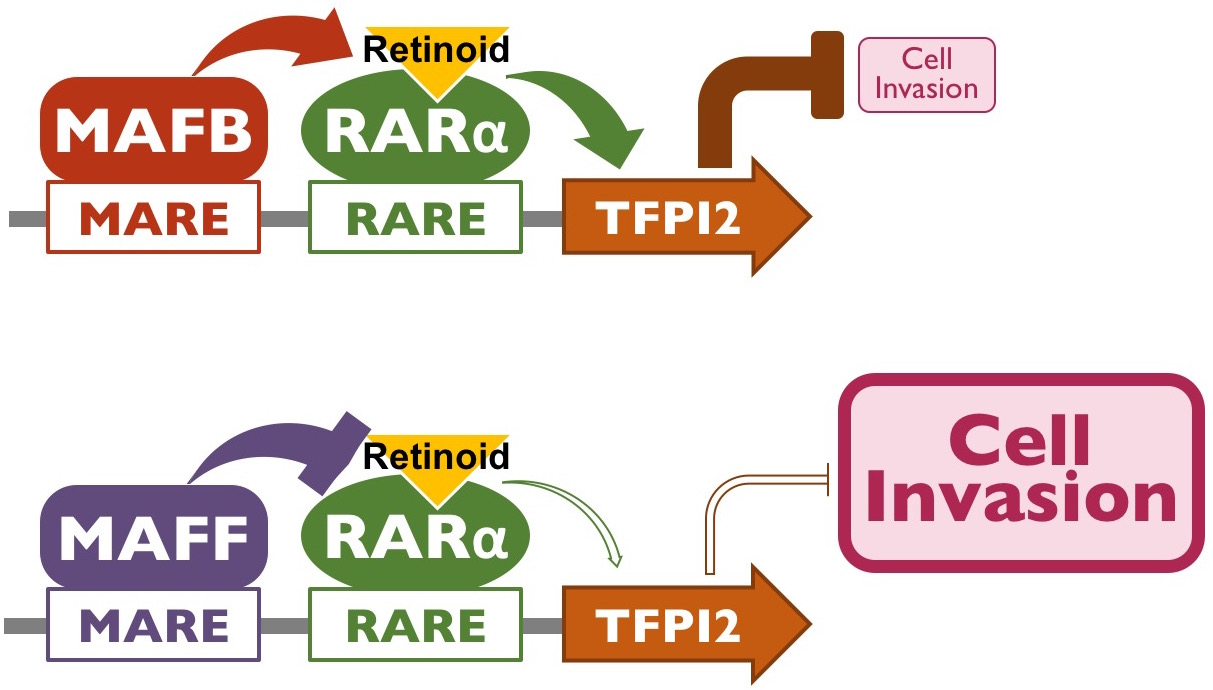{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
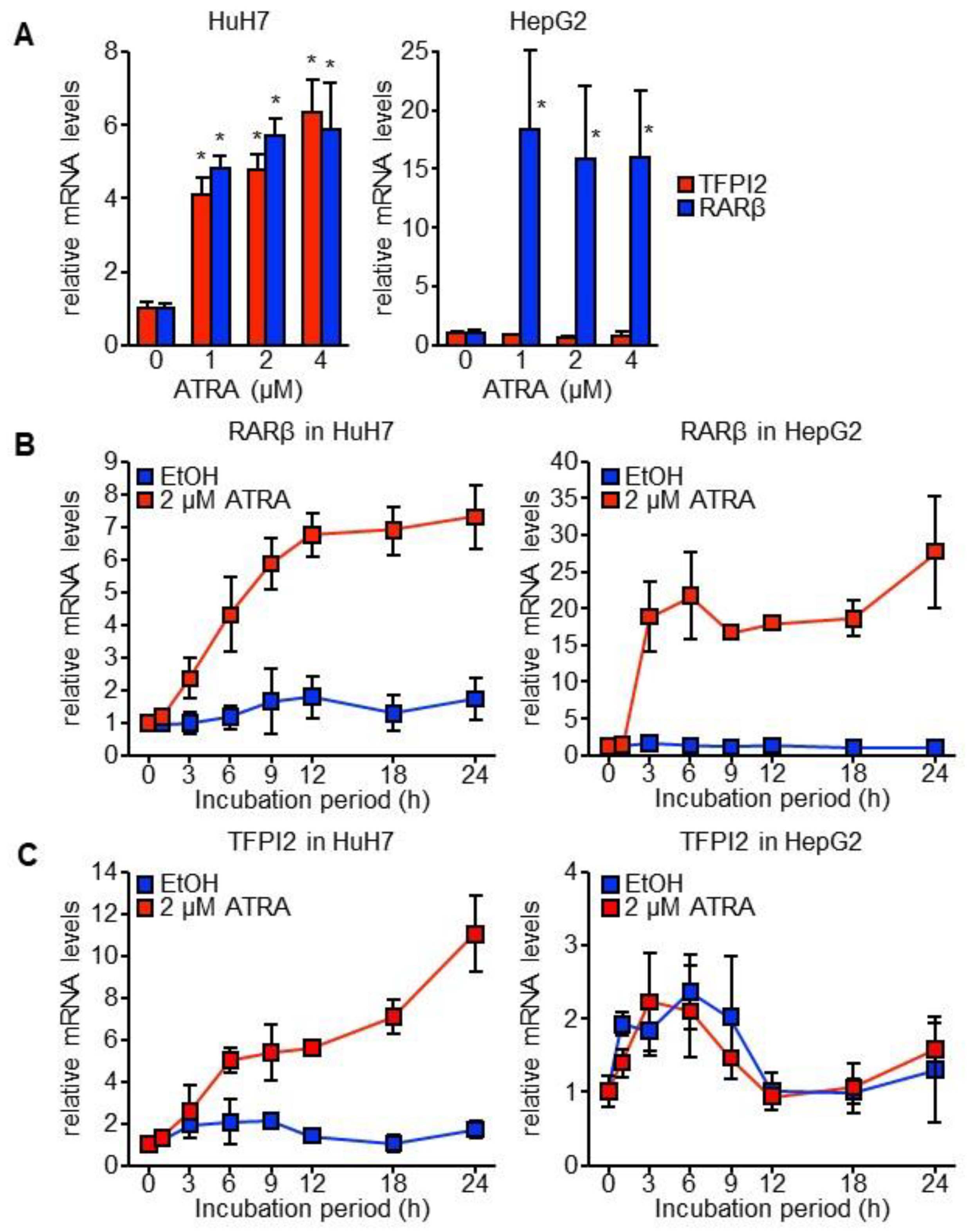
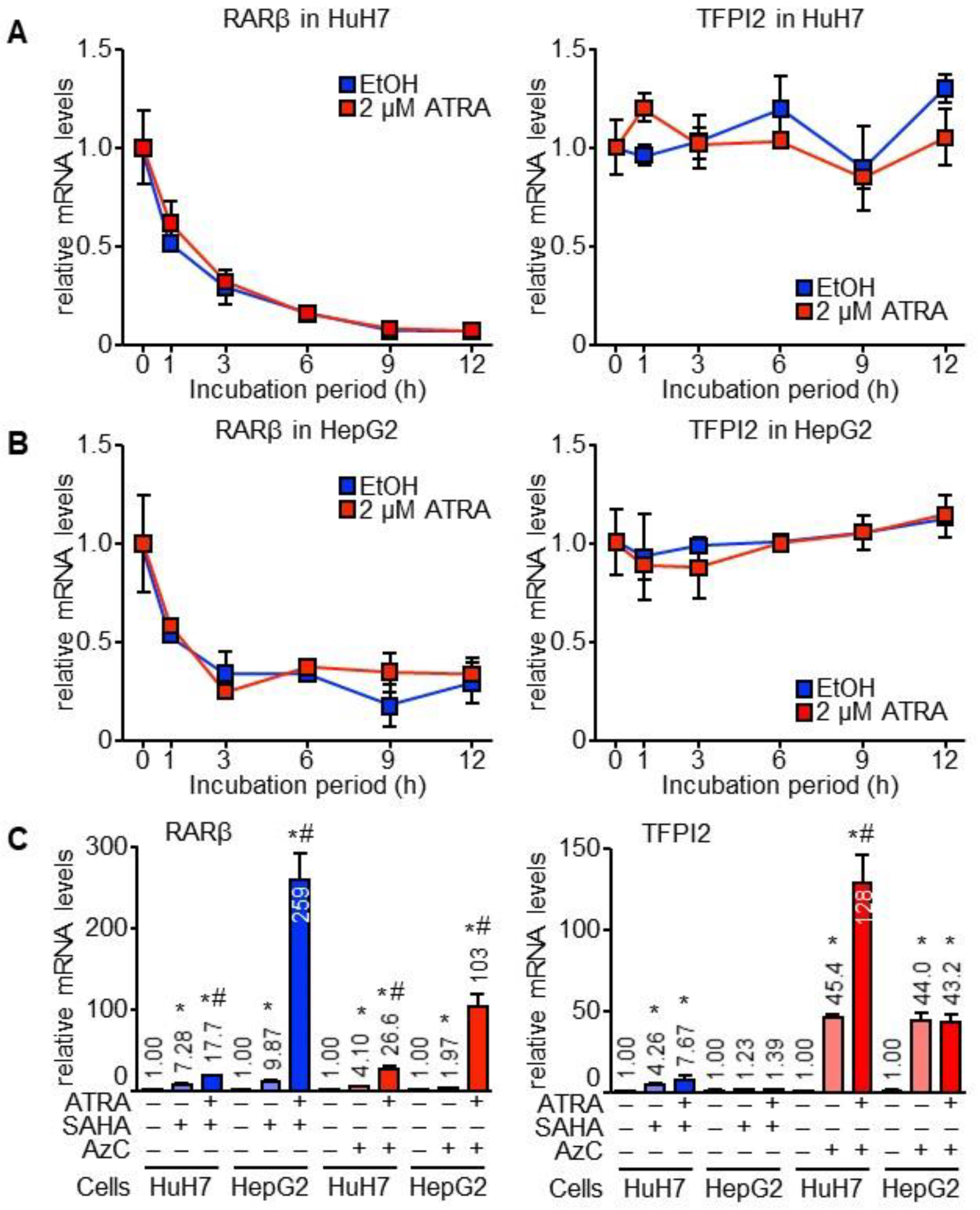
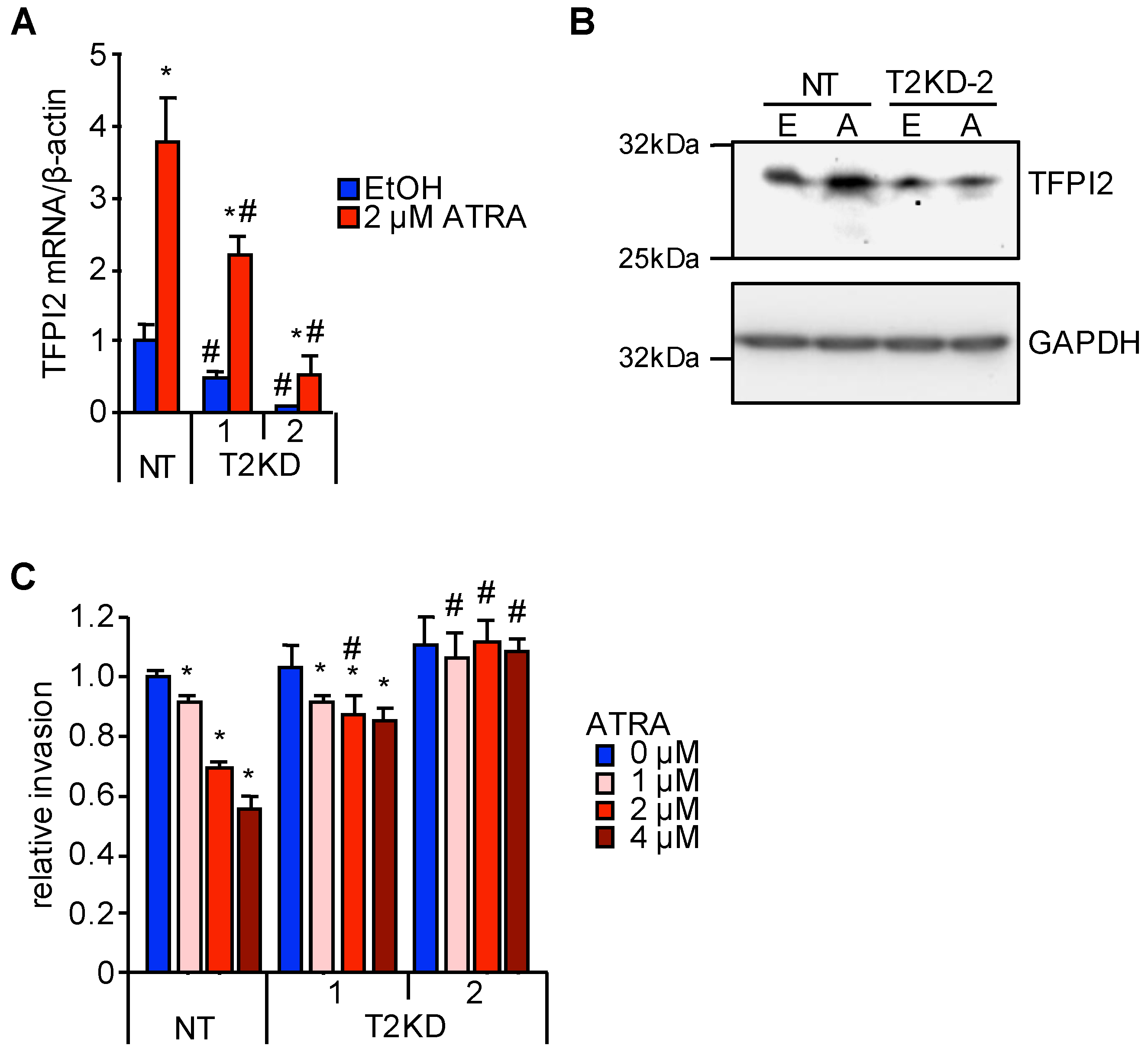
2.1. TFPI2 Is Transcriptionally Regulated by ATRA
2.2. Through TFPI2, ATRA Suppresses HuH7 Cell Invasion
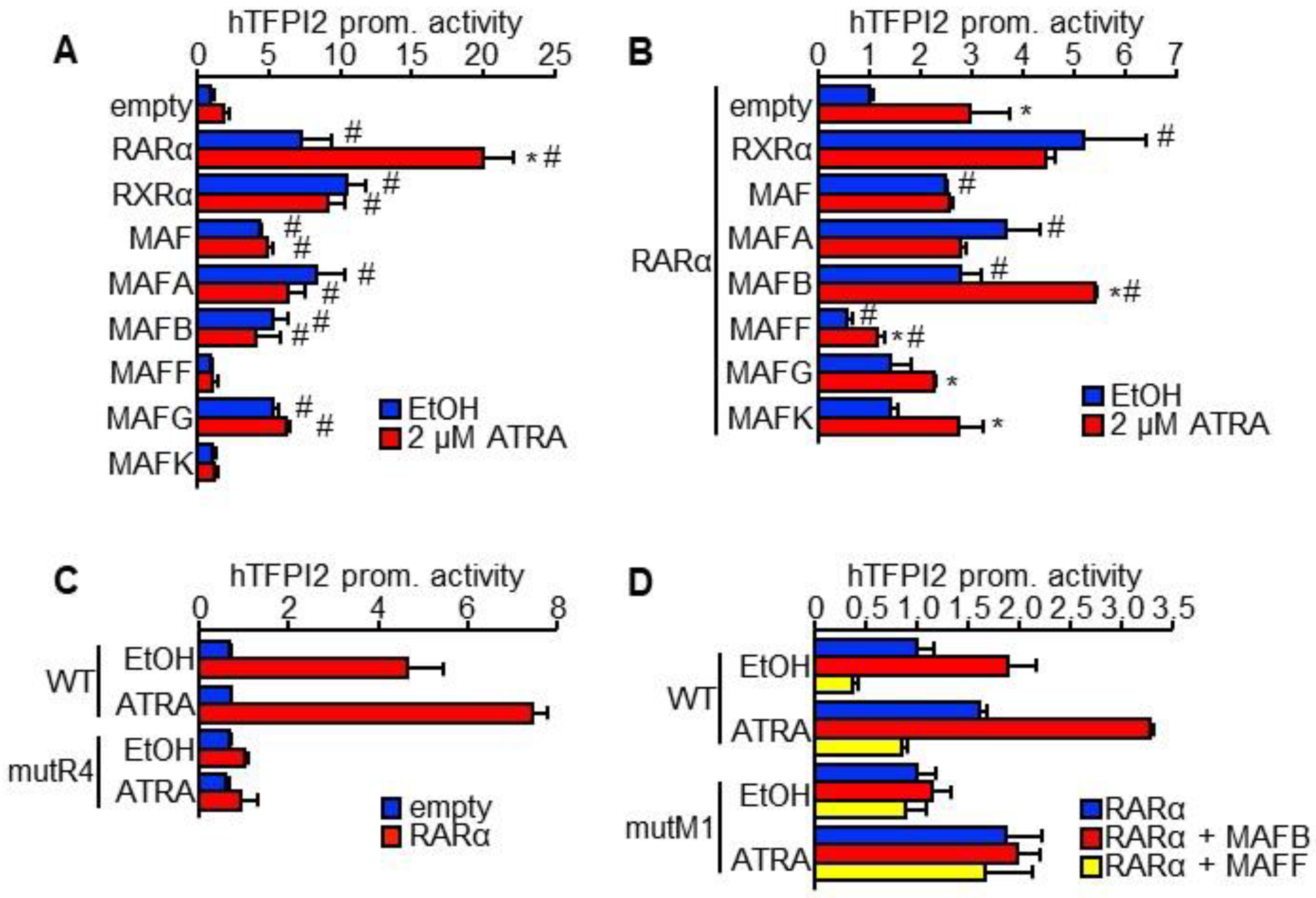
2.3. MAFB and MAFF Modulate the Transactivation Activity of RARα on Human TFPI2 Promoter
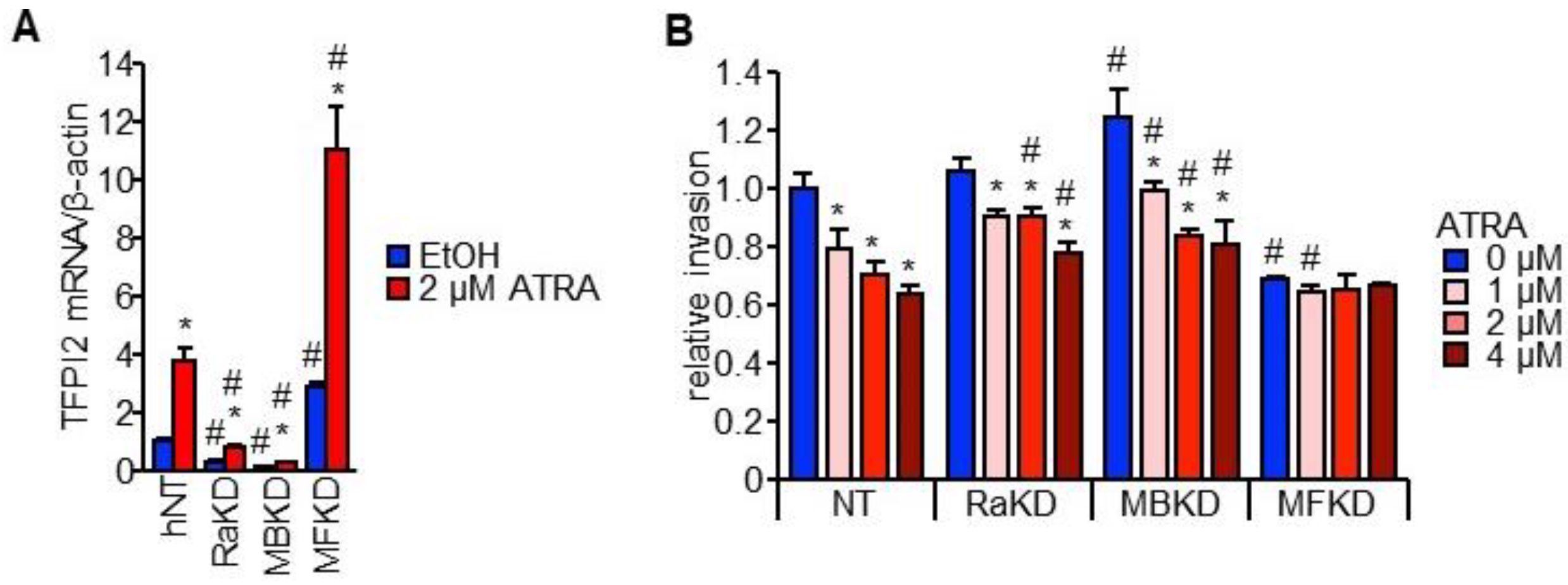
2.4. MAFB, MAFF, and RARα Regulate TFPI2 Expression in HuH7 Cells and Their Invasion
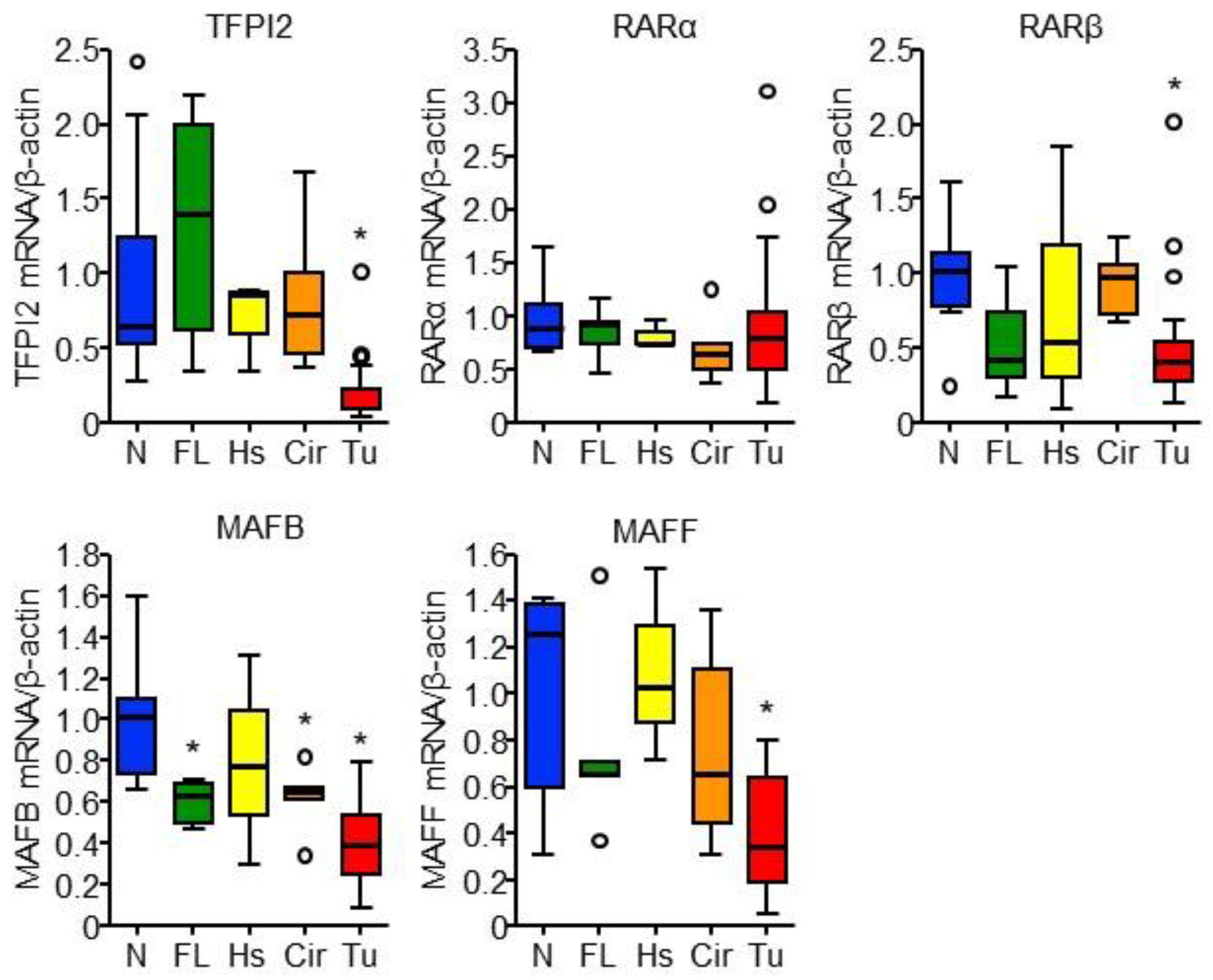
2.5. RARα and MAFB Contribute to the Downregulation of TFPI2 in HCC
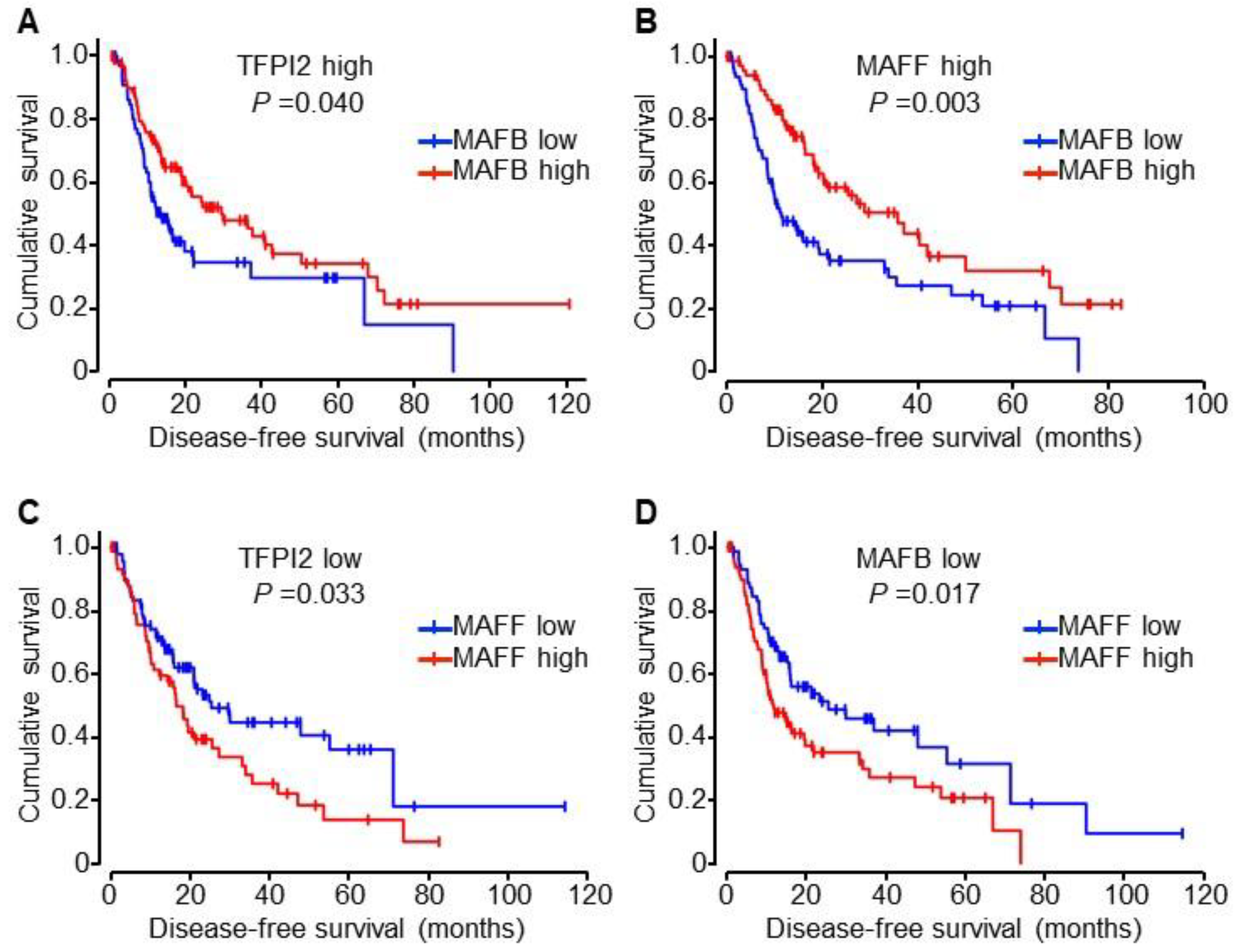
2.6. Involvement of MAFB, MAFF, and TFPI2 in the Progression and Prognosis of HCC
2.7. Biological Significance of MAFB-Regulated ATRA Target Genes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and ATRA Treatments
4.3. Treatment with ActD, AzC, and SAHA
4.4. Establishment of Transgenic Cell Lines
4.5. Determination of mRNA Levels
4.6. Cell Invasion Assay
4.7. Dual Reporter Assay
4.8. Gene Expression Data in Clinical Samples
4.9. Transcription Profiling
4.10. Immunoprecipitation-Western Blotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| RARα | retinoic acid receptor α |
| TFPI2 | tissue factor pathway inhibitor 2 |
| HCC | hepatocellular carcinoma |
| ATRA | all-trans-retinoic acid |
| MAFB | musculoaponeurotic fibrosarcoma oncogene homolog B |
| MAFF | musculoaponeurotic fibrosarcoma oncogene homolog F |
| ActD | actinomycin D |
| AzC | 5-aza-2′-deoxycytidine |
| SAHA | N-hydroxy-N′-phenyloctanediamide |
References
- Zhang, L.; Wang, J.; Liu, L.; Zheng, C.; Wang, Y. Synthesis and Antiproliferative Activity of Novel All-Trans-Retinoic Acid-Podophyllotoxin Conjugate towards Human Gastric Cancer Cells. Molecules 2017, 22, 628. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lu, Y.; Li, D.; Zheng, X.; Lian, J.; Li, S.; Cui, H.; Zhang, L.; Sang, L.; Wang, Y.; et al. All-trans retinoic acid suppresses the angiopoietin-Tie2 pathway and inhibits angiogenesis and metastasis in esophageal squamous cell carcinoma. PLoS ONE 2017, 12, e0174555. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, F.; Zhang, L.; Zhou, Q.; Gui, S.; Wang, Y. A novel all-trans retinoic acid derivative 4-amino-2-trifluoromethyl-phenyl retinate inhibits the proliferation of human hepatocellular carcinoma HepG2 cells by inducing G0/G1 cell cycle arrest and apoptosis via upregulation of p53 and ASPP1 and downregulation of iASPP. Oncol. Rep. 2016, 36, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Orendas, P.; Kubatka, P.; Kajo, K.; Stollarova, N.; Kassayova, M.; Bojkova, B.; Pec, M.; Nosal, V.; Kiskova, T.; Zihlavnikova, K.; et al. Melatonin enhanced bexarotene efficacy in experimental mammary carcinogenesis. Neoplasma 2012, 59, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Ahlersová, E.; Ahlers, I.; Kubatka, P.; Bojková, B.; Môciková, K.; Gajdosová, S.; Onderková, H.M. Melatonin and retinyl acetate as chemopreventives in DMBA-induced mammary carcinogenesis in female Sprague-Dawley rats. Folia. Biol. (Praha) 2000, 46, 69–72. [Google Scholar] [PubMed]
- Hörlein, A.J.; Näär, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Söderström, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Seol, W.; Choi, H.S.; Moore, D.D. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science 1996, 272, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shimada, J.; Shudo, K.; Matsumura, M.; Crippa, M.P.; Kojima, S. Physical interaction between retinoic acid receptor and Sp1: mechanism for induction of urokinase by retinoic acid. Blood 1999, 93, 4264–4276. [Google Scholar] [PubMed]
- Busson, M.; Carazo, A.; Seyer, P.; Grandemange, S.; Casas, F.; Pessemesse, L.; Rouault, J.P.; Wrutniak-Cabello, C.; Cabello, G. Coactivation of nuclear receptors and myogenic factors induces the major BTG1 influence on muscle differentiation. Oncogene 2005, 24, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Zusi, F.C.; Lorenzi, M.V.; Vivat-Hannah, V. Selective retinoids and rexinoids in cancer therapy and chemoprevention. Drug Discov. Today 2002, 7, 1165–1174. [Google Scholar] [CrossRef]
- Sprecher, C.A.; Kisiel, W.; Mathewes, S.; Foster, D.C. Molecular cloning, expression, and partial characterization of a second human tissue-factor-pathway inhibitor. Proc. Natl. Acad. Sci. USA 1994, 91, 3353–3357. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.C.; Sprecher, C.A.; Foster, D.C.; Blumberg, H.; Hamamoto, T.; Kisiel, W. Inhibitory properties of a novel human Kunitz-type protease inhibitor homologous to tissue factor pathway inhibitor. Biochemistry 1996, 35, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.N.; Mohanam, S.; Puppala, A.; Rao, J.S. Regulation of ProMMP-1 and ProMMP-3 activation by tissue factor pathway inhibitor-2/matrix-associated serine protease inhibitor. Biochem. Biophys. Res. Commun. 1999, 255, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Takahashi, C.; Oh, J.; Noda, M. Tissue factor pathway inhibitor-2 suppresses the production of active matrix metalloproteinase-2 and is down-regulated in cells harboring activated ras oncogenes. FEBS Lett. 2000, 481, 31–36. [Google Scholar] [CrossRef]
- Sato, N.; Parker, A.R.; Fukushima, N.; Miyagi, Y.; Iacobuzio-Donahue, C.A.; Eshleman, J.R.; Goggins, M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 2005, 24, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Ng, Y.L.; Lee, J.M.; Wong, C.C.; Cheung, O.F.; Chan, C.Y.; Tung, E.K.; Ching, Y.P.; Ng, I.O. Tissue factor pathway inhibitor-2 as a frequently silenced tumor suppressor gene in hepatocellular carcinoma. Hepatology 2007, 45, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Meex, S.J.; Andreo, U.; Sparks, J.D.; Fisher, E.A. Huh-7 or HepG2 cells: which is the better model for studying human apolipoprotein-B100 assembly and secretion? J. Lipid Res. 2011, 52, 152–158. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Moutier, E.; Ye, T.; Choukrallah, M.A.; Urban, S.; Osz, J.; Chatagnon, A.; Delacroix, L.; Langer, D.; Rochel, N.; Moras, D.; et al. Retinoic acid receptors recognize the mouse genome through binding elements with diverse spacing and topology. J. Biol. Chem. 2012, 287, 26328–26341. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Arenillas, D.; Lim, J.; Swanson, M.I.; Jiang, S.; McCallum, A.; Kirov, S.; Wasserman, W.W. The PAZAR database of gene regulatory information coupled to the ORCA toolkit for the study of regulatory sequences. Nucleic Acids Res. 2007, 37, D54–D60. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Available online: http://cancergenome.nih.gov/ (accessed on 14 December 2016).
- Tsuchiya, H. Kaplan–Meier Analyses of RARα and RARβ in HCC Patients; Tottori University: Tottori, Japan, 2018. [Google Scholar]
- Eychène, A.; Rocques, N.; Pouponnot, C. A new MAFia in cancer. Nat. Rev. Cancer 2008, 8, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Itoh, K.; Motohashi, H.; Hayashi, N.; Matuzaki, Y.; Nakauchi, H.; Nishizawa, M.; Yamamoto, M. Activity and expression of murine small Maf family protein MafK. J. Biol. Chem. 1995, 270, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Ahn, H.; Hagey, L.R.; Romanoski, C.E.; Lee, R.G.; Graham, M.J.; Motohashi, H.; Yamamoto, M.; Edwards, P.A. MAFG is a transcriptional repressor of bile acid synthesis and metabolism. Cell Metab. 2015, 21, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Katsuoka, F.; Motohashi, H.; Engel, J.D.; Yamamoto, M. Embryonic lethality and fetal liver apoptosis in mice lacking all three small Maf proteins. Mol. Cell. Biol. 2012, 32, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Sieweke, M.H.; Tekotte, H.; Frampton, J.; Graf, T. MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell 1996, 85, 49–60. [Google Scholar] [CrossRef]
- He, K.H.; Juhl, K.; Karadimos, M.; El Khattabi, I.; Fitzpatrick, C.; Bonner-Weir, S.; Sharma, A. Differentiation of pancreatic endocrine progenitors reversibly blocked by premature induction of MafA. Dev. Biol. 2014, 385, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Hamada, M.; Morito, N.; Terunuma, T.; Hasegawa, K.; Zhang, C.; Yokomizo, T.; Esaki, R.; Kuroda, E.; Yoh, K.; et al. MafB is essential for renal development and F4/80 expression in macrophages. Mol. Cell. Biol. 2006, 26, 5715–5727. [Google Scholar] [CrossRef] [PubMed]
- Kakehashi, A.; Kato, A.; Ishii, N.; Wei, M.; Morimura, K.; Fukushima, S.; Wanibuchi, H. Valerian inhibits rat hepatocarcinogenesis by activating GABAA receptor-mediated signaling. PLoS ONE 2014, 9, e113610. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Dueñas, C.; González-Herrero, I.; García Cenador, M.B.; García Criado, F.J.; Sánchez-García, I. Loss of p53 exacerbates multiple myeloma phenotype by facilitating the reprogramming of hematopoietic stem/progenitor cells to malignant plasma cells by MafB. Cell Cycle 2012, 11, 3896–3900. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Kataoka, K.; Vogt, P.K. MafA has strong cell transforming ability but is a weak transactivator. Oncogene 2003, 22, 7882–7890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pouponnot, C.; Sii-Felice, K.; Hmitou, I.; Rocques, N.; Lecoin, L.; Druillennec, S.; Felder-Schmittbuhl, M.P.; Eychène, A. Cell context reveals a dual role for Maf in oncogenesis. Oncogene 2006, 25, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Englmeier, U.; Lafon, I.; Sieweke, M.H.; Graf, T. MafB is an inducer of monocytic differentiation. EMBO J. 2000, 19, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.E.; Doggett, N.A.; Albertson, D.; Andaya, A.; Chinnaiyan, A.; van Dekken, H.; Ginzinger, D.; Haqq, C.; James, K.; Kamkar, S.; et al. Integration of high-resolution array comparative genomic hybridization analysis of chromosome 16q with expression array data refines common regions of loss at 16q23-qter and identifies underlying candidate tumor suppressor genes in prostate cancer. Oncogene 2004, 23, 3487–3494. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neaud, V.; Duplantier, J.G.; Mazzocco, C.; Kisiel, W.; Rosenbaum, J. Thrombin up-regulates tissue factor pathway inhibitor-2 synthesis through a cyclooxygenase-2-dependent, epidermal growth factor receptor-independent mechanism. J. Biol. Chem. 2004, 279, 5200–5206. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lin, Y.; Zhang, H.; Liu, J.; Zhang, N.; Li, Y.; Kong, D.; Tang, Q.; Ma, D. Tissue factor pathway inhibitor-2 was repressed by CpG hypermethylation through inhibition of KLF6 binding in highly invasive breast cancer cells. BMC Mol. Biol. 2007, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.H.; He, R.Y.; Chou, J.L.; Chan, M.W.; Li, Y.F.; Tai, C.K. Promoter hypermethylation and silencing of tissue factor pathway inhibitor-2 in oral squamous cell carcinoma. J. Transl. Med. 2014, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.M.; Gao, Y.T.; Ong, C.N.; Ross, R.K.; Yu, M.C. Prediagnostic level of serum retinol in relation to reduced risk of hepatocellular carcinoma. J. Natl. Cancer Inst. 2006, 98, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Ashla, A.A.; Hoshikawa, Y.; Tsuchiya, H.; Hashiguchi, K.; Enjoji, M.; Nakamuta, M.; Taketomi, A.; Maehara, Y.; Shomori, K.; Kurimasa, A.; et al. Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease. Hepatol. Res. 2010, 40, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Izumi, N.; Ikeda, K.; Osaki, Y.; Numata, K.; Ikeda, M.; Kokudo, N.; Imanaka, K.; Nishiguchi, S.; Kondo, S.; et al. Survey of survival among patients with hepatitis C virus-related hepatocellular carcinoma treated with peretinoin, an acyclic retinoid, after the completion of a randomized, placebo-controlled trial. J. Gastroenterol. 2015, 50, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Suto, A.; Iwamoto, T.; Kashiwakuma, D.; Kagami, S.; Suzuki, K.; Takatori, H.; Tamachi, T.; Hirose, K.; Onodera, A.; et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORγt induction as downstream targets of Stat3. J. Exp. Med. 2014, 211, 1857–1874. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, M.; Sone, H.; Amemiya, M.; Katoh, Y.; Isogai, M.; Shimano, H.; Yamada, N.; Takahashi, S. Mouse MafA, homologue of zebrafish somite Maf 1, contributes to the specific transcriptional activity through the insulin promoter. Biochem. Biophys. Res. Commun. 2003, 312, 831–842. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hamada, M.; Nakamura, M.; Tran, M.T.; Moriguchi, T.; Hong, C.; Ohsumi, T.; Dinh, T.T.; Kusakabe, M.; Hattori, M.; Katsumata, T.; et al. MafB promotes atherosclerosis by inhibiting foam-cell apoptosis. Nat. Commun. 2014, 5, 3147. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuchiya, H.; Oura, S. Involvement of MAFB and MAFF in Retinoid-Mediated Suppression of Hepatocellular Carcinoma Invasion. Int. J. Mol. Sci. 2018, 19, 1450. https://doi.org/10.3390/ijms19051450
Tsuchiya H, Oura S. Involvement of MAFB and MAFF in Retinoid-Mediated Suppression of Hepatocellular Carcinoma Invasion. International Journal of Molecular Sciences. 2018; 19(5):1450. https://doi.org/10.3390/ijms19051450
Chicago/Turabian StyleTsuchiya, Hiroyuki, and Seiya Oura. 2018. "Involvement of MAFB and MAFF in Retinoid-Mediated Suppression of Hepatocellular Carcinoma Invasion" International Journal of Molecular Sciences 19, no. 5: 1450. https://doi.org/10.3390/ijms19051450
APA StyleTsuchiya, H., & Oura, S. (2018). Involvement of MAFB and MAFF in Retinoid-Mediated Suppression of Hepatocellular Carcinoma Invasion. International Journal of Molecular Sciences, 19(5), 1450. https://doi.org/10.3390/ijms19051450