A New Venue of TNF Targeting



Abstract
1. Introduction
2. Biology
3. TNF in Health and Disease
3.1. Homeostatic Functions of TNF in Immunity
3.2. Systemic Inflammation
3.2.1. TNF: The Master Regulator of Inflammation
3.2.2. Differential Roles for TNFR1 and TNFR2 in Sepsis
3.3. Autoimmunity
3.3.1. Implications of TNF Signaling
3.3.2. Receptor-Dependent Roles for TNF in Autoimmunity
3.4. The Role of TNF in Neurodegenerative Diseases
3.4.1. Differential Roles of TNFR1 and TNFR2 in Neuronal Health and Disease
3.4.2. TNF and Its Receptors in Multiple Sclerosis
3.4.3. TNF Involvement in Alzheimer’s Disease
3.4.4. TNF in Parkinson’s Disease
4. TNF Inhibitors
4.1. Approved TNF Inhibitors
4.2. Mechanisms of Action of TNF Inhibitors
4.3. Pitfalls of TNF Inhibitors
4.3.1. High Costs
4.3.2. Clinical Response
4.3.3. Increased Susceptibility to Infection and Malignancies
4.3.4. Demyelinating Disease and Other Neurological Side Effects
4.3.5. Paradoxical Side Effects
5. Other Anti-TNF and TNF-Modulating Drugs
5.1. TNF Inhibitors
5.2. TNF Modulators
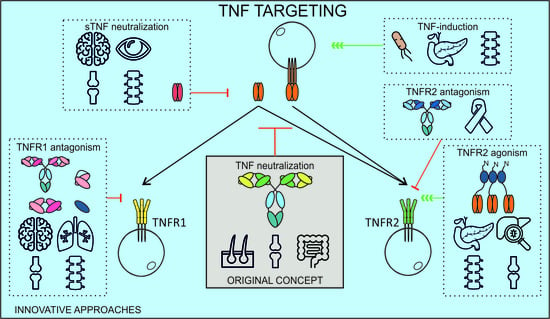
6. A New Chapter of Inventive TNF Manipulating Approaches
6.1. Selective TNFR1 Targeting
6.2. Selective TNFR2 Targeting
6.3. TNF-Inducing Vaccines
6.4. Selective Targeting of sTNF
6.5. Cell-Type Restricted TNF(R) Targeting
6.6. Multispecific Approaches
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| ADA | Anti-drug antibody |
| ADAM | A disintegrin and metalloproteinase |
| ADCC | Antibody-dependent cellular cytotoxicity |
| ALS | Amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| APC | Antigen-presenting cells |
| AS | Ankylosing spondylitis |
| Aβ(O) | (oligomerized) Amyloid beta |
| BACE1 | Beta-secretase 1 |
| BBB | Blood–brain barrier |
| BCG | Bacillus Calmette-Guérin |
| CAIA | Collagen antibody-induced arthritis |
| cAMP | Cyclic AMP |
| CASP | Colon ascendens stent peritonitis |
| CD | Crohn’s disease |
| CDC | Cell-dependent cytotoxicity |
| CIA | Collagen-induced arthritis |
| CLP | Cecal ligation and puncture |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CSF | Cerebrospinal fluid |
| DC | Dendritic cell |
| DD | Death domain |
| DILE | Drug-induced lupus erythematosus |
| DSS | Dextran sodium sulfate |
| EAE | Experimental autoimmune encephalomyelitis |
| EMA | European Medicine Agency |
| FADD | Fas-associated death domain |
| FcγR | Fcγ-receptor |
| FDA | Food and Drug Administration |
| FDC | Follicular dendritic cell |
| GC | Germinal center |
| GVHD | Graft-versus-host disease |
| HT | 5-hydroxytryptamine |
| hTNF | Human TNF |
| ip | Intraperitoneal |
| IBD | Inflammatory bowel disease |
| icv | Intracerebroventricular |
| IEC | Intestinal epithelial cells |
| IFN | Interferon |
| IFNAR | Interferon-α receptor |
| Ig | Immunoglobulin |
| IL | Interleukin |
| JIA | Juvenile idiopathic arthritis |
| KO | Knockout |
| LPS | Lipopolysaccharide |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| LT-α | Lymphotoxin-α |
| mAb | Monoclonal antibody |
| MDSC | Myeloid-derived suppressor cells |
| MMP | Matrix metalloproteinase |
| MOF | Multiple organ failure |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | Multiple sclerosis |
| MTX | Methotrexate |
| Nb | Nanobody |
| NEC | Necrotizing enterocolitis |
| NK | Natural killer |
| NOD | Non-obese diabetic |
| OLG | Oligodendrocytes |
| OPC | Oligodendrocyte precursor cell |
| PD | Parkinson’s disease |
| PEG | Polyethylene glycol |
| PLAD | Pre-ligand assembly domain |
| PPMS | Primary progressive MS |
| PsA | Psoriatic arthritis |
| QoL | Quality-of-life |
| RA | Rheumatoid arthritis |
| ROS | Reactive oxygen species |
| RRMS | Relapsing-remitting MS |
| scFv | Single-chain variable fragment |
| SIRS | Systemic inflammatory response syndrome |
| SLE | Systemic lupus erythematosus |
| sTNF(R) | Soluble TNF(R) |
| TACE | TNF-α converting enzyme |
| TDM | Therapeutic drug monitoring |
| TIMP | Tissue inhibitor of metalloproteinase |
| tmTNF | Transmembrane TNF |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| TRADD | TNFR1-associated death domain |
| TRAF(2) | TNF receptor-associated factor (2) |
| TRAPS | TNF receptor-associated periodic syndrome |
| Tregs | Regulatory T cells |
| UC | Ulcerative colitis |
| VHH | Variable domain of heavy-chain only Abs |
| VNAR | Variable new antigen receptor |
| WT | Wild type |
References
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 Years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J. Tumor necrosis factor (TNF). Science 1985, 230, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Creaven, P.J.; Plager, J.E.; Dupere, S.; Huben, R.P.; Takita, H.; Mittelman, A.; Proefrock, A. Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother. Pharmacol. 1987, 20, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, B.; Kurzrock, R.; Talpaz, M.; Blick, M.; Saks, S.; Gutterman, J.U. A phase I trial of intravenously-administered recombinant tumor necrosis factor-alpha in cancer patients. J. Clin. Oncol. 1988, 6, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Zhou, S.; Diaz, L.A., Jr.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Naismith, J.H.; Sprang, S.R. Modularity in the TNF-receptor family. Trends Biochem. Sci. 1998, 23, 74–79. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Gearing, A.J.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L.; et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. Targeting TNFR2, an immune checkpoint stimulator and oncoprotein, is a promising treatment for cancer. Sci. Signal. 2017, 10, eaal2328. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal. 2017, 10, eaaf8608. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Ayres, T.M.; Wong, G.H.; Goeddel, D.V. A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74, 845–853. [Google Scholar] [CrossRef]
- Faustman, D.L.; Davis, M. TNF receptor 2 and disease: Autoimmunity and regenerative medicine. Front. Immunol. 2013, 4, 478. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Nunes, T.; Bernardazzi, C.; de Souza, H.S. Cell death and inflammatory bowel diseases: Apoptosis, necrosis, and autophagy in the intestinal epithelium. BioMed Res. Int. 2014, 2014, 218493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.A.; Lehman, J.; Roderick, J.E.; Martinez-Marin, D.; Zelic, M.; Doran, C.; Hermance, N.; Lyle, S.; Pasparakis, M.; Fitzgerald, K.A.; et al. Dendritic cell RIPK1 maintains immune homeostasis by preventing inflammation and autoimmunity. J. Immunol. 2018, 200, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Lohden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Wallach, D.; Engelmann, H.; Nophar, Y.; Aderka, D.; Kemper, O.; Hornik, V.; Holtmann, H.; Brakebusch, C. Soluble and cell surface receptors for tumor necrosis factor. Agents Actions Suppl. 1991, 35, 51–57. [Google Scholar] [PubMed]
- Aderka, D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996, 7, 231–240. [Google Scholar] [CrossRef]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kda TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Mootoo, A.; Stylianou, E.; Arias, M.A.; Reljic, R. TNF-alpha in tuberculosis: A cytokine with a split personality. Inflamm. Allergy Drug Targets 2009, 8, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kollias, G.; Douni, E.; Kassiotis, G.; Kontoyiannis, D. The function of tumour necrosis factor and receptors in models of multi-organ inflammation, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Ann. Rheum. Dis. 1999, 58 (Suppl. 1), I32–I39. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Alexopoulou, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNF alpha-deficient mice: A critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.W.; Dunn, A.; Grail, D.; Inglese, M.; Noguchi, Y.; Richards, E.; Jungbluth, A.; Wada, H.; Moore, M.; Williamson, B.; et al. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8093–8098. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, S.; Rickelt, S.; Soriano, M.E.; Schips, T.G.; Maier, H.J.; Davos, C.H.; Varela, A.; Kaklamanis, L.; Mann, D.L.; Capetanaki, Y. Tumor necrosis factor-alpha confers cardioprotection through ectopic expression of keratins K8 and K18. Nat. Med. 2015, 21, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Bluml, S.; Binder, N.B.; Niederreiter, B.; Polzer, K.; Hayer, S.; Tauber, S.; Schett, G.; Scheinecker, C.; Kollias, G.; Selzer, E.; et al. Antiinflammatory effects of tumor necrosis factor on hematopoietic cells in a murine model of erosive arthritis. Arthritis Rheum. 2010, 62, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Loetscher, H.; Stueber, D.; Gehr, G.; Lesslauer, W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993, 177, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.; Maier, O. Targeting sTNF/TNFR1 signaling as a new therapeutic strategy. Antibodies 2015, 4, 48–70. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.; Marino, M.W.; Brown, N.; Abel, B.; Bekker, L.G.; Quesniaux, V.J.; Fick, L.; Ryffel, B. Correction of defective host response to Mycobacterium Bovis BCG infection in TNF-deficient mice by bone marrow transplantation. Lab. Investig. 2000, 80, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Allie, N.; Grivennikov, S.I.; Keeton, R.; Hsu, N.J.; Bourigault, M.L.; Court, N.; Fremond, C.; Yeremeev, V.; Shebzukhov, Y.; Ryffel, B.; et al. Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci. Rep. 2013, 3, 1809. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Lesslauer, W.; Lotscher, H.; Lang, Y.; Koebel, P.; Kontgen, F.; Althage, A.; Zinkernagel, R.; Steinmetz, M.; Bluethmann, H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Mackay, F.; Bluethmann, H.; Zinkernagel, R. Phenotypic analysis of TNFR1-deficient mice and characterization of TNFR1-deficient fibroblasts in vitro. Circ. Shock 1994, 44, 51–56. [Google Scholar] [PubMed]
- Flynn, J.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schreiber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef]
- Garcia, I.; Olleros, M.L.; Quesniaux, V.F.; Jacobs, M.; Allie, N.; Nedospasov, S.A.; Szymkowski, D.E.; Ryffel, B. Roles of soluble and membrane TNF and related ligands in mycobacterial infections: Effects of selective and non-selective TNF inhibitors during infection. Adv. Exp. Med. Biol. 2011, 691, 187–201. [Google Scholar] [PubMed]
- Segueni, N.; Benmerzoug, S.; Rose, S.; Gauthier, A.; Bourigault, M.L.; Reverchon, F.; Philippeau, A.; Erard, F.; Le Bert, M.; Bouscayrol, H.; et al. Innate myeloid cell TNFR1 mediates first line defence against primary Mycobacterium tuberculosis infection. Sci. Rep. 2016, 6, 22454. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.G.; Tompkins, R.G.; Gelfand, J.A.; Michie, H.R.; Stanford, G.G.; van der Meer, J.W.; Endres, S.; Lonnemann, G.; Corsetti, J.; Chernow, B.; et al. Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J. Infect. Dis. 1990, 161, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Gogos, C.A.; Drosou, E.; Bassaris, H.P.; Skoutelis, A. Pro-versus anti-inflammatory cytokine profile in patients with severe sepsis: A marker for prognosis and future therapeutic options. J. Infect. Dis. 2000, 181, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Feezor, R.J.; Oberholzer, C.; Baker, H.V.; Novick, D.; Rubinstein, M.; Moldawer, L.L.; Pribble, J.; Souza, S.; Dinarello, C.A.; Ertel, W.; et al. Molecular characterization of the acute inflammatory response to infections with gram-negative versus gram-positive bacteria. Infect. Immun. 2003, 71, 5803–5813. [Google Scholar] [CrossRef] [PubMed]
- Schulte, W.; Bernhagen, J.; Bucala, R. Cytokines in sepsis: Potent immunoregulators and potential therapeutic targets—An updated view. Mediat. Inflamm. 2013, 2013, 165974. [Google Scholar] [CrossRef] [PubMed]
- Rigato, O.; Ujvari, S.; Castelo, A.; Salomao, R. Tumor necrosis factor alpha (TNF-alpha) and sepsis: Evidence for a role in host defense. Infection 1996, 24, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Kothari, N.; Bogra, J.; Abbas, H.; Kohli, M.; Malik, A.; Kothari, D.; Srivastava, S.; Singh, P.K. Tumor necrosis factor gene polymorphism results in high TNF level in sepsis and septic shock. Cytokine 2013, 61, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, E.; Sarikcioglu, A.; Aksaray, N.; Seydaoglu, G.; Seyhun, Y.; Yaman, A. Role of procalcitonin, C-reactive protein, interleukin-6, interleukin-8 and tumor necrosis factor-alpha in the diagnosis of neonatal sepsis. Turk. J. Pediatr. 2007, 49, 7–20. [Google Scholar] [PubMed]
- Abraham, E.; Laterre, P.F.; Garbino, J.; Pingleton, S.; Butler, T.; Dugernier, T.; Margolis, B.; Kudsk, K.; Zimmerli, W.; Anderson, P.; et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: A randomized, double-blind, placebo-controlled, multicenter phase iii trial with 1342 patients. Crit. Care Med. 2001, 29, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Glauser, M.P.; Butler, T.; Garbino, J.; Gelmont, D.; Laterre, P.F.; Kudsk, K.; Bruining, H.A.; Otto, C.; Tobin, E.; et al. P55 tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 study group. JAMA 1997, 277, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Wunderink, R.; Silverman, H.; Perl, T.M.; Nasraway, S.; Levy, H.; Bone, R.; Wenzel, R.P.; Balk, R.; Allred, R.; et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAB sepsis study group. JAMA 1995, 273, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 1985, 229, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Newham, P.; Ross, D.; Ceuppens, P.; Das, S.; Yates, J.W.; Betts, C.; Reens, J.; Randall, K.J.; Knight, R.; McKay, J.S. Determination of the safety and efficacy of therapeutic neutralization of tumor necrosis factor-α (TNF-α) using azd9773, an anti-TNF-α immune fab, in murine clp sepsis. Inflamm. Res. 2014, 63, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, K.; Menges, T.; Gardlund, B.; Harm Zwaveling, J.; Smithes, M.; Vincent, J.L.; Tellado, J.M.; Salgado-Remigio, A.; Zimlichman, R.; Withington, S.; et al. Randomized, placebo-controlled trial of the anti-tumor necrosis factor antibody fragment afelimomab in hyperinflammatory response during severe sepsis: The ramses study. Crit. Care Med. 2001, 29, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, M.K.; Bolgos, G.; Miller, C.; Nguyen, D.T.; DeForge, L.E.; Remick, D.G. Anti-tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J. Immunol. 1992, 148, 2724–2730. [Google Scholar] [PubMed]
- Secher, T.; Vasseur, V.; Poisson, D.M.; Mitchell, J.A.; Cunha, F.Q.; Alves-Filho, J.C.; Ryffel, B. Crucial role of TNF receptors 1 and 2 in the control of polymicrobial sepsis. J. Immunol. 2009, 182, 7855–7864. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Han, M.; Yi, R.; Kwon, S.; Dai, C.; Wang, R. Anti-TNF-α therapy for patients with sepsis: A systematic meta-analysis. Int. J. Clin. Pract. 2014, 68, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. The anti-inflammatory effect of exercise: Its role in diabetes and cardiovascular disease control. Essays Biochem. 2006, 42, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, K.; Matsuyama, T.; Kündig, T.M.; Wakeham, A.; Kishihara, K.; Shahinian, A.; Wiegmann, K.; Ohashi, P.S.; Krönke, M.; Mak, T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. Monocytogenes infection. Cell 1993, 73, 457–467. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Hauwermeiren, F.; Lodens, S.; De Rycke, R.; Van Wonterghem, E.; Staes, A.; Gevaert, K.; Lopez-Otin, C.; Libert, C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol. Med. 2013, 5, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J.; Torrance, D.S.; Stocking, K.L.; Glaccum, M.B.; Otten, C.; Willis, C.R.; Charrier, K.; Morrissey, P.J.; Ware, C.B.; Mohler, K.M. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 1998, 160, 943–952. [Google Scholar] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Watson, A.J.; Frey, M.R.; Miguel, J.C.; Burkitt, M.D.; Sutton, R.; Hughes, K.R.; Hall, L.J.; Caamaño, J.H.; et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis. Models Mech. 2013, 6, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Van Hauwermeiren, F.; Armaka, M.; Karagianni, N.; Kranidioti, K.; Vandenbroucke, R.E.; Loges, S.; Van Roy, M.; Staelens, J.; Puimege, L.; Palagani, A.; et al. Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J. Clin. Investig. 2013, 123, 2590–2603. [Google Scholar] [CrossRef] [PubMed]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Grine, L.; Lodens, S.; Van Wonterghem, E.; De Rycke, R.; De Geest, N.; Hassan, B.; Libert, C. TNFR1-induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol. 2015, 8, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Van Ryckeghem, S.; Vandewalle, J.; Ballegeer, M.; Van Wonterghem, E.; Eggermont, M.; Decruyenaere, J.; De Bus, L.; Libert, C.; Vandenbroucke, R.E. Simultaneous inhibition of tumor necrosis factor receptor 1 and matrix metalloproteinase 8 completely protects against acute inflammation and sepsis. Crit. Care Med. 2018, 46, e67–e75. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Lint, P.; Demeestere, D.; Van Wonterghem, E.; Vanlaere, I.; Puimege, L.; Van Hauwermeiren, F.; De Rycke, R.; Mc Guire, C.; et al. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-csf barrier contributes to lethality during systemic inflammatory diseases. J. Neurosci. 2012, 32, 9805–9816. [Google Scholar] [CrossRef] [PubMed]
- Tauber, S.C.; Eiffert, H.; Bruck, W.; Nau, R. Septic encephalopathy and septic encephalitis. Expert Rev. Anti Infect. Ther. 2017, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Jacob, A.; Cunningham, P.; Hensley, L.; Quigg, R.J. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem. Int. 2008, 52, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Calsavara, A.C.; Soriani, F.M.; Vieira, L.Q.; Costa, P.A.; Rachid, M.A.; Teixeira, A.L. TNFR1 absence protects against memory deficit induced by sepsis possibly through over-expression of hippocampal BDNF. Metab. Brain Dis. 2015, 30, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Zantl, N.; Uebe, A.; Neumann, B.; Wagner, H.; Siewert, J.R.; Holzmann, B.; Heidecke, C.D.; Pfeffer, K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect. Immun. 1998, 66, 2300–2309. [Google Scholar] [PubMed]
- Hildebrand, F.; Pape, H.C.; Hoevel, P.; Krettek, C.; van Griensven, M. The importance of systemic cytokines in the pathogenesis of polymicrobial sepsis and dehydroepiandrosterone treatment in a rodent model. Shock 2003, 20, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Ebach, D.R.; Riehl, T.E.; Stenson, W.F. Opposing effects of tumor necrosis factor receptor 1 and 2 in sepsis due to cecal ligation and puncture. Shock 2005, 23, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G.; Call, D.R.; Ebong, S.J.; Newcomb, D.E.; Nybom, P.; Nemzek, J.A.; Bolgos, G.E. Combination immunotherapy with soluble tumor necrosis factor receptors plus interleukin 1 receptor antagonist decreases sepsis mortality. Crit. Care Med. 2001, 29, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bonnet, M.C.; Ulvmar, M.H.; Wolk, K.; Karagianni, N.; Witte, E.; Uthoff-Hachenberg, C.; Renauld, J.C.; Kollias, G.; Toftgard, R.; et al. Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin-24-dependent psoriasis-like skin inflammation in mice. Immunity 2013, 39, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Reinecker, H.C.; Steffen, M.; Witthoeft, T.; Pflueger, I.; Schreiber, S.; MacDermott, R.P.; Raedler, A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin. Exp. Immunol. 1993, 94, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Kollias, G. Fibroblast biology. Synovial fibroblasts in rheumatoid arthritis: Leading role or chorus line? Arthritis Res. 2000, 2, 342–343. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mori, L.; Iselin, S.; De Libero, G.; Lesslauer, W. Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG1-treated and TNFR1-deficient mice. J. Immunol. 1996, 157, 3178–3182. [Google Scholar] [PubMed]
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s disease cA2 study group. N. Engl. J. Med. 1997, 337, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF Au-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [PubMed]
- Li, G.; Wu, Y.; Jia, H.; Tang, L.; Huang, R.; Peng, Y.; Zhang, Y. Establishment and evaluation of a transgenic mouse model of arthritis induced by overexpressing human tumor necrosis factor alpha. Biol. Open 2016, 5, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Kollias, G.; Kontoyiannis, D. Role of TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002, 13, 315–321. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Noti, M.; Corazza, N.; Mueller, C.; Berger, B.; Brunner, T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J. Exp. Med. 2010, 207, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Chi, H.H.; Rajaiah, R.; Moudgil, K.D. Exogenous tumour necrosis factor alpha induces suppression of autoimmune arthritis. Arthritis Res. Ther. 2008, 10, R38. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Seino, H.; Abo, T.; Tanaka, S.; Shintani, S.; Ohta, S.; Tamura, K.; Sawai, T.; Nobunaga, T.; Oteki, T.; et al. Recombinant human tumor necrosis factor alpha suppresses autoimmune diabetes in nonobese diabetic mice. J. Clin. Investig. 1989, 84, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Grewal, K.D.; Wong, F.S.; Picarella, D.E.; Janeway, C.A., Jr.; Flavell, R.A. Local expression of transgene encoded TNF alpha in islets prevents autoimmune diabetes in nonobese diabetic (NOD) mice by preventing the development of auto-reactive islet-specific T cells. J. Exp. Med. 1996, 184, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.O.; McDevitt, H.O. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature 1988, 331, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Kollias, G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur. J. Immunol. 2000, 30, 2038–2047. [Google Scholar] [CrossRef]
- Kassiotis, G.; Kollias, G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: Implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 2001, 193, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Libert, C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011, 22, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Ho, A.; Koarada, S.; Morito, F.; Ushiyama, O.; Suzuki, N.; Kikuchi, Y.; Ohta, A.; Mak, T.W.; Nagasawa, K. Collagen-induced arthritis in TNF receptor-1-deficient mice: TNF receptor-2 can modulate arthritis in the absence of TNF receptor-1. Clin. Immunol. 2001, 99, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y.; Erdmann, J.; Alexopoulou, L.; Kollias, G.; Ross, F.P.; Teitelbaum, S.L. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 2000, 275, 27307–27310. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Baumel, M.; Mannel, D.N.; Howard, O.M.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007, 179, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Subleski, J.J.; Hamano, R.; Howard, O.M.; Wiltrout, R.H.; Oppenheim, J.J. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FoxP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 2010, 40, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011, 585, 3611–3618. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, B.; Chen, X.; Oppenheim, J.J.; Baeyens, A.; Gregoire, S.; Chader, D.; Gorochov, G.; Miyara, M.; Salomon, B.L. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med. 2016, 22, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.M.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3+ conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Balancing TNFR1 and TNFR2 jointly for joint inflammation. Arthritis Rheumatol. 2014, 66, 2657–2660. [Google Scholar] [CrossRef] [PubMed]
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.; Stoop, A.A.; Williams, R.O. Selective TNFR1 blockade is anti-inflammatory and reveals an immunoregulatory role for TNFR2. Arthritis Rheumatol. 2014, 66, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Housley, W.J.; Adams, C.O.; Nichols, F.C.; Puddington, L.; Lingenheld, E.G.; Zhu, L.; Rajan, T.V.; Clark, R.B. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J. Immunol. 2011, 186, 6779–6787. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, X.; Zhou, Q.; Howard, O.M.; Netea, M.G.; Oppenheim, J.J. TNFR2 is critical for the stabilization of the CD4+FoxP3+ regulatory t. Cell phenotype in the inflammatory environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Boden, E.K.; Snapper, S.B. Regulatory T cells in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2008, 24, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Pierik, M.; Vermeire, S.; Steen, K.V.; Joossens, S.; Claessens, G.; Vlietinck, R.; Rutgeerts, P. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment. Pharmacol. Ther. 2004, 20, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Bae, S.C.; Lee, Y.H. Associations between functional TNFR2 196 m/r polymorphisms and susceptibility to rheumatoid arthritis: A meta-analysis. Rheumatol. Int. 2014, 34, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Anderson, S.K. Association of TNFRSF1B promoter polymorphisms with human disease: Further studies examining T-regulatory cells are required. Front. Immunol. 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C.; Mattson, M.P. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse 2000, 35, 151–159. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Sama, D.M.; Mohmmad Abdul, H.; Furman, J.L.; Artiushin, I.A.; Szymkowski, D.E.; Scheff, S.W.; Norris, C.M. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and cA2+ dysregulation in aged rats. PLoS ONE 2012, 7, e38170. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.G.; Banks, W.A.; Kastin, A.J. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993, 47, 169–176. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed]
- Hofman, F.M.; Hinton, D.R.; Johnson, K.; Merrill, J.E. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med. 1989, 170, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Kuno, R.; Wang, J.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Autocrine activation of microglia by tumor necrosis factor-alpha. J. Neuroimmunol. 2005, 162, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNF-alpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-d-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa b pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, L.; Lindholm, K.; Konishi, Y.; Yue, X.; Hampel, H.; Zhang, D.; Shen, Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J. Neurosci. 2004, 24, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lindholm, K.; Konishi, Y.; Li, R.; Shen, Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J. Neurosci. 2002, 22, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Stacey, D.; Redlich, R.; Buschel, A.; Opel, N.; Grotegerd, D.; Zaremba, D.; Dohm, K.; Burger, C.; Meinert, S.L.; Forster, K.; et al. TNF receptors 1 and 2 exert distinct region-specific effects on striatal and hippocampal grey matter volumes (VBM) in healthy adults. Genes Brain Behav. 2017, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Palmer, T.D. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav. Immun. 2013, 30, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Zoecklein, L.; Papke, L.; Gamez, J.; Denic, A.; Macura, S.; Howe, C. Tumor necrosis factor alpha is reparative via TNFR2 in the hippocampus and via TNFR1 in the striatum after virus-induced encephalitis. Brain Pathol. 2009, 19, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Dellarole, A.; Morton, P.; Brambilla, R.; Walters, W.; Summers, S.; Bernardes, D.; Grilli, M.; Bethea, J.R. Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav. Immun. 2014, 41, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Heldmann, U.; Thored, P.; Claasen, J.H.; Arvidsson, A.; Kokaia, Z.; Lindvall, O. TNF-alpha antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp. Neurol. 2005, 196, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.J.; Boling, W.; Kindy, M.S.; Peschon, J.; Kraemer, P.J.; Carpenter, M.K.; Holtsberg, F.W.; Mattson, M.P. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 1996, 2, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, P.B.; Provitera, V.; De Rosa, T.; Tartaglia, G.; Gorga, F.; Perrella, O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: A correlation with clinical activity. Immunopharmacol. Immunotoxicol. 1998, 20, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Doolittle, T.H.; Lincoln, R.; Brown, R.H.; Dinarello, C.A. Cytokine accumulations in CSF of multiple sclerosis patients: Frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology 1990, 40, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Almoallim, H.; Al-Ghamdi, Y.; Almaghrabi, H.; Alyasi, O. Anti-tumor necrosis factor-α induced systemic lupus erythematosus. Open Rheumatol. J. 2012, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Tack, C.J.; Kleijwegt, F.S.; Van Riel, P.L.; Roep, B.O. Development of type 1 diabetes in a patient treated with anti-TNF-alpha therapy for active rheumatoid arthritis. Diabetologia 2009, 52, 1442–1444. [Google Scholar] [CrossRef] [PubMed]
- Probert, L.; Akassoglou, K.; Pasparakis, M.; Kontogeorgos, G.; Kollias, G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1995, 92, 11294–11298. [Google Scholar] [CrossRef] [PubMed]
- Akassoglou, K.; Probert, L.; Kontogeorgos, G.; Kollias, G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J. Immunol. 1997, 158, 438–445. [Google Scholar] [PubMed]
- Zajicek, J.P.; Wing, M.; Scolding, N.J.; Compston, D.A. Interactions between oligodendrocytes and microglia. A major role for complement and tumour necrosis factor in oligodendrocyte adherence and killing. Brain J. Neurol. 1992, 115 Pt 6, 1611–1631. [Google Scholar] [CrossRef]
- Kruglov, A.A.; Lampropoulou, V.; Fillatreau, S.; Nedospasov, S.A. Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J. Immunol. 2011, 187, 5660–5670. [Google Scholar] [CrossRef] [PubMed]
- Ruddle, N.H.; Bergman, C.M.; McGrath, K.M.; Lingenheld, E.G.; Grunnet, M.L.; Padula, S.J.; Clark, R.B. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 1990, 172, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Bauer, J.; Akassoglou, K.; Lassmann, H.; Kollias, G.; Probert, L. A tumor necrosis factor-induced model of human primary demyelinating diseases develops in immunodeficient mice. Eur. J. Immunol. 1999, 29, 912–917. [Google Scholar] [CrossRef]
- Frei, K.; Eugster, H.P.; Bopst, M.; Constantinescu, C.S.; Lavi, E.; Fontana, A. Tumor necrosis factor alpha and lymphotoxin alpha are not required for induction of acute experimental autoimmune encephalomyelitis. J. Exp. Med. 1997, 185, 2177–2182. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; von Blomberg, B.M.; Woody, J.N.; Hartung, H.P.; Polman, C.H. Increased mri activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Arnason, B.G.W. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. The lenercept multiple sclerosis study group and the university of british columbia ms/mri analysis group. Neurology 1999, 53, 457–465. [Google Scholar]
- D’Souza, S.D.; Bonetti, B.; Balasingam, V.; Cashman, N.R.; Barker, P.A.; Troutt, A.B.; Raine, C.S.; Antel, J.P. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J. Exp. Med. 1996, 184, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.W.; Raine, C.S. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann. Neurol. 1988, 23, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Raine, C.S.; Farooq, M.; Norton, W.T.; Brosnan, C.F. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J. Immunol. 1991, 147, 1522–1529. [Google Scholar] [PubMed]
- Akassoglou, K.; Bauer, J.; Kassiotis, G.; Pasparakis, M.; Lassmann, H.; Kollias, G.; Probert, L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: Models for multiple sclerosis with primary oligodendrogliopathy. Am. J. Pathol. 1998, 153, 801–813. [Google Scholar] [CrossRef]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1a as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Eugster, H.P.; Frei, K.; Bachmann, R.; Bluethmann, H.; Lassmann, H.; Fontana, A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 1999, 29, 626–632. [Google Scholar] [CrossRef]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35–55)-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Kranidioti, K.; Xanthoulea, S.; Denis, M.; Kotanidou, A.; Douni, E.; Blackshear, P.J.; Kontoyiannis, D.L.; Kollias, G. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur. J. Immunol. 2006, 36, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Novrup, H.G.; Bracchi-Ricard, V.; Ellman, D.G.; Ricard, J.; Jain, A.; Runko, E.; Lyck, L.; Yli-Karjanmaa, M.; Szymkowski, D.E.; Pearse, D.D.; et al. Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J. Neuroinflamm. 2014, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-κB. Brain J. Neurol. 2011, 134, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain J. Neurol. 2011, 134, 2736–2754. [Google Scholar] [CrossRef] [PubMed]
- Evangelidou, M.; Karamita, M.; Vamvakas, S.S.; Szymkowski, D.E.; Probert, L. Altered expression of oligodendrocyte and neuronal marker genes predicts the clinical onset of autoimmune encephalomyelitis and indicates the effectiveness of multiple sclerosis-directed therapeutics. J. Immunol. 2014, 192, 4122–4133. [Google Scholar] [CrossRef] [PubMed]
- Karamita, M.; Barnum, C.; Mobius, W.; Tansey, M.G.; Szymkowski, D.E.; Lassmann, H.; Probert, L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight 2017, 2, e87455. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Abe, Y.; Kamada, H.; Shibata, H.; Kayamuro, H.; Inoue, M.; Kawara, T.; Arita, S.; Furuya, T.; Yamashita, T.; et al. Therapeutic effect of pegylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J. Control. Release 2011, 149, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, e90117. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Van Ryckeghem, S.; Van Imschoot, G.; De Rycke, R.; Toussaint, W.; Vanhoutte, L.; Vanhove, C.; De Vos, F.; Vandenbroucke, R.E.; Libert, C. TNFR1 inhibition with a nanobody protects against EAE development in mice. Sci. Rep. 2017, 7, 13646. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNF-alpha impairs memory via astrocyte signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fischer, R.; Naude, P.J.; Maier, O.; Nyakas, C.; Duffey, M.; Van der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Fischer, R.; Agresti, C.; Pfizenmaier, K. TNF receptor 2 protects oligodendrocyte progenitor cells against oxidative stress. Biochem. Biophys. Res. Commun. 2013, 440, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Motti, D.; Karmally, S.; Szymkowski, D.E.; Lambertsen, K.L.; Bethea, J.R.; Brambilla, R. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J. Neurosci. 2016, 36, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Wajant, H.; Kontermann, R.; Pfizenmaier, K.; Maier, O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 2014, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Buc, M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediat. Inflamm. 2013, 2013, 963748. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, N.; Papadopoulos, D.; Denis, M.C.; Mitsikostas, D.D.; Kollias, G. TNFR2 on non-haematopoietic cells is required for FoxP3+ treg-cell function and disease suppression in eae. Eur. J. Immunol. 2012, 42, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Arico, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, R.; Eugster, H.P.; Frei, K.; Fontana, A.; Lassmann, H. Impairment of TNF-receptor-1 signaling but not fas signaling diminishes T-cell apoptosis in myelin oligodendrocyte glycoprotein peptide-induced chronic demyelinating autoimmune encephalomyelitis in mice. Am. J. Pathol. 1999, 154, 1417–1422. [Google Scholar] [CrossRef]
- Ban, L.; Zhang, J.; Wang, L.; Kuhtreiber, W.; Burger, D.; Faustman, D.L. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc. Natl. Acad. Sci. USA 2008, 105, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Kartalou, G.I.; Zenelak, S.; Georgopoulos, S. Peripheral tumor necrosis factor-alpha (TNF-alpha) modulates amyloid pathology by regulating blood-derived immune cells and glial response in the brain of AD/TNF transgenic mice. J. Neurosci. 2017, 37, 5155–5171. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Gorle, N.; Vandendriessche, C.; Balusu, S.; Brkic, M.; Van Cauwenberghe, C.; Van Imschoot, G.; Van Wonterghem, E.; De Rycke, R.; Kremer, A.; et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, P.; Xie, J.; Staufenbiel, M.; Li, R.; Shen, Y. Genetic deletion of TNFRII gene enhances the Alzheimer-like pathology in an APP transgenic mouse model via reduction of phosphorylated IκBα. Hum. Mol. Genet. 2014, 23, 4906–4918. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, R.; Shiosaki, K. Inhibition of p75 tumor necrosis factor receptor by antisense oligonucleotides increases hypoxic injury and beta-amyloid toxicity in human neuronal cell line. J. Biol. Chem. 1997, 272, 3550–3553. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.L.; Narrow, W.C.; Mastrangelo, M.A.; Olschowka, J.A.; O’Banion, M.K.; Bowers, W.J. Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic Alzheimer disease mice leads to significant modulation of amyloid- and tau-related pathologies. Am. J. Pathol. 2013, 182, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, J.; Rowan, M.J.; Anwyl, R. Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur. J. Neurosci. 2005, 22, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Leng, A.; Mura, A.; Feldon, J.; Ferger, B. Tumor necrosis factor-alpha receptor ablation in a chronic MPTP mouse model of Parkinson’s disease. Neurosci. Lett. 2005, 375, 107–111. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Ruhn, K.A.; Martinez, T.N.; McAlpine, F.E.; Blesch, A.; Tansey, M.G. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiParkinsonian rats. Mol. Ther. 2008, 16, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiParkinsonian rats. J. Parkinsons Dis. 2014, 4, 349–360. [Google Scholar] [PubMed]
- Sullivan, P.G.; Bruce-Keller, A.J.; Rabchevsky, A.G.; Christakos, S.; Clair, D.K.; Mattson, M.P.; Scheff, S.W. Exacerbation of damage and altered NF-κB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J. Neurosci. 1999, 19, 6248–6256. [Google Scholar] [CrossRef] [PubMed]
- Longhi, L.; Ortolano, F.; Zanier, E.R.; Perego, C.; Stocchetti, N.; De Simoni, M.G. Effect of traumatic brain injury on cognitive function in mice lacking p55 and p75 tumor necrosis factor receptors. Acta Neurochir. Suppl. 2008, 102, 409–413. [Google Scholar] [PubMed]
- Longhi, L.; Perego, C.; Ortolano, F.; Aresi, S.; Fuma galli, S.; Zanier, E.R.; Stocchetti, N.; De Simoni, M.G. Tumor necrosis factor in traumatic brain injury: Effects of genetic deletion of p55 or p75 receptor. J. Cereb. Blood Flow Metab. 2013, 33, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Gary, D.S.; Bruce-Keller, A.J.; Kindy, M.S.; Mattson, M.P. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 1998, 18, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef] [PubMed]
- Balosso, S.; Ravizza, T.; Perego, C.; Peschon, J.; Campbell, I.L.; De Simoni, M.G.; Vezzani, A. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann. Neurol. 2005, 57, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Balosso, S.; Ravizza, T.; Pierucci, M.; Calcagno, E.; Invernizzi, R.; Di Giovanni, G.; Esposito, E.; Vezzani, A. Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: Implications for seizure susceptibility. Neuroscience 2009, 161, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Schafers, M.; Schmidt, C.; Vogel, C.; Toyka, K.V.; Sommer, C. Tumor necrosis factor-alpha (TNF) regulates the expression of ICAM-1 predominantly through TNF receptor 1 after chronic constriction injury of mouse sciatic nerve. Acta Neuropathol. 2002, 104, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Stallforth, S.; Sommer, C. Altered pain behavior and regeneration after nerve injury in TNF receptor deficient mice. J. Peripher. Nerv. Syst. JPNS 2006, 11, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E. Tumour necrosis factor modulation for treatment of Alzheimer’s disease: Rationale and current evidence. CNS Drugs 2009, 23, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial tnfalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Zenelak, S.; Georgopoulos, S. Genetic deletion of tumor necrosis factor-alpha attenuates amyloid-beta production and decreases amyloid plaque formation and glial response in the 5xfad model of Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2017, 60, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Marx, F.; Steiner, E.; Hartmann, T.; Grubeck-Loebenstein, B. TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999, 13, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, J.; Landreth, G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiyota, T.; Horiba, M.; Buescher, J.L.; Walsh, S.M.; Gendelman, H.E.; Ikezu, T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in swedish mutant APP transgenic mice. Am. J. Pathol. 2007, 170, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 2011, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Janelsins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.M.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3XTG-AD mice. Am. J. Pathol. 2008, 173, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.C.; Kane, M.; Ghimire, S.; Gautam, S.; Gui, J. Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: A nested case-control analysis. CNS Drugs 2016, 30, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.M.; Lin, M.T.; Larson, E.B.; Maezawa, I.; Tseng, L.H.; Edwards, K.L.; Schellenberg, G.D.; Hansen, J.A.; Kukull, W.A.; Jin, L.W. Tumor necrosis factor alpha and interleukin 10 promoter region polymorphisms and risk of late-onset Alzheimer disease. Arch. Neurol. 2006, 63, 1165–1169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lewis, M.; Tartaglia, L.A.; Lee, A.; Bennett, G.L.; Rice, G.C.; Wong, G.H.; Chen, E.Y.; Goeddel, D.V. Cloning and expression of cdnas for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA 1991, 88, 2830–2834. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.Q.; Zhang, Q.Q.; Zhang, Y.D.; Xu, J. Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11C-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Choi, S.M.; Jho, J.; Park, M.S.; Kang, J.; Park, S.J.; Ryu, J.H.; Jo, J.; Kim, H.H.; Kim, B.C. Infliximab ameliorates AD-associated object recognition memory impairment. Behav. Brain Res. 2016, 311, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, F.; Vernay, A.; Leuba, G.; Schenk, F. Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolism. Brain Res. Bull. 2009, 80, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Knox, J.; Chang, J.; Derbedrossian, A.; Vasilevko, V.; Cribbs, D.; Boado, R.J.; Pardridge, W.M.; Sumbria, R.K. Blood-brain barrier penetrating biologic TNF-alpha inhibitor for Alzheimer’s disease. Mol. Pharm. 2017, 14, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Puntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-alpha modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed 2006, 8, 25. [Google Scholar] [PubMed]
- Tobinick, E.L.; Gross, H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J. Neuroinflamm. 2008, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Wang, B.R.; Jiang, W.W.; Chen, J.; Zhu, Y.W.; Zhong, L.L.; Zhang, Y.D.; Xu, J. Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J. Am. Geriatr. Soc. 2011, 59, 1142–1144. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, S.; Huber, L.; Stein, R.; Schmid, K.; Swick, A.; Wand, P.; Brody, M.; Strum, S.; Joyal, S.V. Open label, crossover, pilot study to assess the efficacy and safety of perispinal administration of etanercept (enbrel®) in combination with nutritional supplements versus nutritional supplements alone in subjects with mild to moderate Alzheimer’s disease receiving standard care. FASEB J. 2016, 30 (Suppl. 1), Ib296. [Google Scholar]
- Cheng, X.; Yang, L.; He, P.; Li, R.; Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J. Alzheimer’s Dis. JAD 2010, 19, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Cribbs, D.H.; Anderson, A.J.; Cummings, B.J.; Su, J.H.; Wasserman, A.J.; Cotman, C.W. The induction of the TNF-alpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem. Res. 2003, 28, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorle, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid beta oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.; Herrera, A.J.; Venero, J.L.; Santiago, M.; De Pablos, R.M.; Villaran, R.F.; Espinosa-Oliva, A.M.; Arguelles, S.; Sarmiento, M.; Delgado-Cortes, M.J.; et al. Peripheral inflammation increases the damage in animal models of nigrostriatal dopaminergic neurodegeneration: Possible implication in Parkinson’s disease incidence. Parkinson’s Dis. 2011, 2011, 393769. [Google Scholar] [CrossRef] [PubMed]
- Kouchaki, E.; Kakhaki, R.D.; Tamtaji, O.R.; Dadgostar, E.; Behnam, M.; Nikoueinejad, H.; Akbari, H. Increased serum levels of TNF-alpha and decreased serum levels of IL-27 in patients with Parkinson disease and their correlation with disease severity. Clin. Neurol. Neurosurg. 2018, 166, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Lindenau, J.D.; Altmann, V.; Schumacher-Schuh, A.F.; Rieder, C.R.; Hutz, M.H. Tumor necrosis factor alpha polymorphisms are associated with Parkinson’s disease age at onset. Neurosci. Lett. 2017, 658, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from Parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-alpha. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Rousselet, E.; Callebert, J.; Parain, K.; Joubert, C.; Hunot, S.; Hartmann, A.; Jacque, C.; Perez-Diaz, F.; Cohen-Salmon, C.; Launay, J.M.; et al. Role of TNF-alpha receptors in mice intoxicated with the Parkinsonian toxin MPTP. Exp. Neurol. 2002, 177, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Chertoff, M.; Di Paolo, N.; Schoeneberg, A.; Depino, A.; Ferrari, C.; Wurst, W.; Pfizenmaier, K.; Eisel, U.; Pitossi, F. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor alpha in the nigrostriatal dopaminergic circuit of adult mice. Exp. Neurol. 2011, 227, 237–251. [Google Scholar] [CrossRef] [PubMed]
- De Lella Ezcurra, A.L.; Chertoff, M.; Ferrari, C.; Graciarena, M.; Pitossi, F. Chronic expression of low levels of tumor necrosis factor-alpha in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol. Dis. 2010, 37, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Gemma, C.; Catlow, B.; Cole, M.; Hudson, C.; Samec, A.; Shah, N.; Vila, J.; Bachstetter, A.; Bickford, P.C. Early inhibition of tnfalpha increases 6-hydroxydopamine-induced striatal degeneration. Brain Res. 2007, 1147, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.H.; Sumbria, R.; Hui, E.K.; Lu, J.Z.; Boado, R.J.; Pardridge, W.M. Neuroprotection with a brain-penetrating biologic tumor necrosis factor inhibitor. J. Pharmacol. Exp. Ther. 2011, 339, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Casabona, J.C.; Puntel, M.; Pitossi, F.J. Interleukin-1beta and tumor necrosis factor-alpha: Reliable targets for protective therapies in Parkinson’s disease? Front. Cell. Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Cerami, A. The value of failure: The discovery of TNF and its natural inhibitor erythropoietin. J. Intern. Med. 2011, 269, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Anzueto, A.; Gutierrez, G.; Tessler, S.; San Pedro, G.; Wunderink, R.; Dal Nogare, A.; Nasraway, S.; Berman, S.; Cooney, R.; et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. Norasept II study group. Lancet 1998, 351, 929–933. [Google Scholar] [CrossRef]
- Williams, R.O.; Feldmann, M.; Maini, R.N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 1992, 89, 9784–9788. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Katsikis, P.; Brennan, F.M.; Walker, J.; Bijl, H.; Ghrayeb, J.; et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993, 36, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.A.; Eisen, D.B. Treatment of hidradenitis suppurativa with biologic medications. J. Am. Acad. Dermatol. 2015, 73, S82–S88. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.H.; Racewicz, A.; Brzezicki, J.; Yatsyshyn, R.; Arteaga, E.T.; Baranauskaite, A.; Abud-Mendoza, C.; Navarra, S.; Kadinov, V.; Sariego, I.G.; et al. A phase III randomized study to evaluate the efficacy and safety of CT-p13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the planetra study. Arthritis Res. Ther. 2016, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Von Richter, O.; Skerjanec, A.; Afonso, M.; Sanguino Heinrich, S.; Poetzl, J.; Woehling, H.; Velinova, M.; Koch, A.; Kollins, D.; Macke, L.; et al. Gp2015, a proposed etanercept biosimilar: Pharmacokinetic similarity to its reference product and comparison of its autoinjector device with prefilled syringes. Br. J. Clin. Pharmacol. 2017, 83, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549. [Google Scholar] [CrossRef]
- Yoo, D.H.; Prodanovic, N.; Jaworski, J.; Miranda, P.; Ramiterre, E.; Lanzon, A.; Baranauskaite, A.; Wiland, P.; Abud-Mendoza, C.; Oparanov, B.; et al. Efficacy and safety of CT-p13 (biosimilar infliximab) in patients with rheumatoid arthritis: Comparison between switching from reference infliximab to CT-p13 and continuing CT-p13 in the planetra extension study. Ann. Rheum. Dis. 2017, 76, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.K.; Shih, D.Q.; Chen, G.C. Insights on the use of biosimilars in the treatment of inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, S.; Mrowietz, U.; Augustin, M.; Ralph von, K.; Enk, A.; Stromer, K.; Schon, M.P.; Radtke, M.A. Biosimilars in dermatology—Theory becomes reality. J. Ger. Soc. Dermatol. JDDG 2018, 16, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Marotte, H.; Cimaz, R. Etanercept—TNF receptor and IgG1 Fc fusion protein: Is it different from other TNF blockers? Expert Opin. Biol. Ther. 2014, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Thaci, D.; Gerdes, S.; Arenberger, P.; Pulka, G.; Kingo, K.; Weglowska, J.; Hattebuhr, N.; Poetzl, J.; Woehling, H.; et al. The EGALITY study: A confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. The originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br. J. Dermatol. 2017, 176, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Keystone, E.C.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Teoh, L.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Khaliq-Kareemi, M.; Lawrance, I.C.; Thomsen, O.O.; Hanauer, S.B.; McColm, J.; Bloomfield, R.; Sandborn, W.J. Maintenance therapy with certolizumab pegol for Crohn’s disease. N. Engl. J. Med. 2007, 357, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Hutas, G. Golimumab as the first monthly subcutaneous fully human anti-TNF-alpha antibody in the treatment of inflammatory arthropathies. Immunotherapy 2010, 2, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.S.; Hartman, D.; Lemos, B.R.; Erlich, E.C.; Spence, S.; Kennedy, S.; Ptak, T.; Pruitt, R.; Vermeire, S.; Fox, B.S. AVX-470, an orally delivered anti-tumour necrosis factor antibody for treatment of active ulcerative colitis: Results of a first-in-human trial. J. Crohn’s Colitis 2016, 10, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Hartman, D.S.; Tracey, D.E.; Lemos, B.R.; Erlich, E.C.; Burton, R.E.; Keane, D.M.; Patel, R.; Kim, S.; Bhol, K.C.; Harris, M.S.; et al. Effects of AVX-470, an oral, locally acting anti-tumour necrosis factor antibody, on tissue biomarkers in patients with active ulcerative colitis. J. Crohn’s Colitis 2016, 10, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Lichtenstein, G.; Radford-Smith, G.; Patel, J.; Innes, A. CDP571, a humanized monoclonal antibody to tumour necrosis factor-alpha, for steroid-dependent Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Aliment. Pharmacol. Ther. 2006, 23, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Nayiager, S.; Louw, I.; Rojkovich, B.; Fu, C.; Udata, C. A multiple ascending dose/proof of concept study of ATN-103 (ozoralizumab) in rheumatoid arthritis subjects on a background of methotrexate. Arthritis Rheum. 2011, 63, 2630. [Google Scholar]
- Beirnaert, E.; Desmyter, A.; Spinelli, S.; Lauwereys, M.; Aarden, L.; Dreier, T.; Loris, R.; Silence, K.; Pollet, C.; Cambillau, C.; et al. Bivalent Llama single-domain antibody fragments against tumor necrosis factor have picomolar potencies due to intramolecular interactions. Front. Immunol. 2017, 8, 867. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.K., 3rd. PEGylated recombinant human soluble tumour necrosis factor receptor type I (r-Hu-sTNF-RI): Novel high affinity TNF receptor designed for chronic inflammatory diseases. Ann. Rheum. Dis. 1999, 58 (Suppl. 1), I73–I81. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.W.; Feige, U.; Bendele, A.M.; Martin, S.W.; Edwards, C.K., 3rd. Treatment of rheumatoid arthritis with PEGylated recombinant human soluble tumour necrosis factor receptor type I: A clinical update. Ann. Rheum. Dis. 2000, 59 (Suppl. 1), i41–i43. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, G.; Gu, C.; Chen, B.; Chen, A.; Li, H.; Gao, B.; Liang, C.; Wu, J.; Yang, T.; et al. T0001, a variant of TNFR2-Fc fusion protein, exhibits improved Fc effector functions through increased binding to membrane-bound TNF-alpha. PLoS ONE 2017, 12, e0177891. [Google Scholar]
- Rice, T.W.; Wheeler, A.P.; Morris, P.E.; Paz, H.L.; Russell, J.A.; Edens, T.R.; Bernard, G.R. Safety and efficacy of affinity-purified, anti-tumor necrosis factor-alpha, ovine fab for injection (CytoFab) in severe sepsis. Crit. Care Med. 2006, 34, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Furst, D.E.; Fleischmann, R.; Kopp, E.; Schiff, M.; Edwards, C.K.; Solinger, A.; Macri, M.; Grp, S. A phase 2 dose-finding study of pegylated recombinant methionyl human soluble tumor necrosis factor type I in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 2303–2310. [Google Scholar] [PubMed]
- Durez, P.; Vandepapeliere, P.; Miranda, P.; Toncheva, A.; Berman, A.; Kehler, T.; Mociran, E.; Fautrel, B.; Mariette, X.; Dhellin, O.; et al. Therapeutic vaccination with TNF-kinoid in TNF antagonist-resistant rheumatoid arthritis: A phase II randomized, controlled clinical trial. PLoS ONE 2014, 9, e113465. [Google Scholar] [CrossRef] [PubMed]
- Vandepapeliere, P.; Malen, F.; Rogler, G.; Van der Bijl, A.; Kruger, F.C.; Kruger, D.W.; Grouard-Vogel, G.; Dhellin, O.; Fanget, B.; Michetti, P.F. Safety, immunogenicity and clinical phase I-II results of TNFalpha-kinoid immunotherapeutic in Crohn’s disease patients. Gastroenterology 2011, 140, S-123. [Google Scholar] [CrossRef]
- Spohn, G.; Guler, R.; Johansen, P.; Keller, I.; Jacobs, M.; Beck, M.; Rohner, F.; Bauer, M.; Dietmeier, K.; Kundig, T.M.; et al. A virus-like particle-based vaccine selectively targeting soluble TNF-alpha protects from arthritis without inducing reactivation of latent tuberculosis. J. Immunol. 2007, 178, 7450–7457. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, R.K.; Boado, R.J.; Pardridge, W.M. Brain protection from stroke with intravenous tnfalpha decoy receptor-trojan horse fusion protein. J. Cereb. Blood Flow Metab. 2012, 32, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Olesen, C.M.; Coskun, M.; Peyrin-Biroulet, L.; Nielsen, O.H. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol. Ther. 2016, 159, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.J.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14+ macrophages. Gastroenterology 2011, 141, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.D.; Wildenberg, M.E.; van den Brink, G.R. Mechanism of action of anti-TNF therapy in inflammatory bowel disease. J. Crohn’s Colitis 2016, 10, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Scallon, B.; Cai, A.; Solowski, N.; Rosenberg, A.; Song, X.Y.; Shealy, D.; Wagner, C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J. Pharmacol. Exp. Ther. 2002, 301, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, A.; Fossati, G.; Bergin, M.; Stephens, P.; Stephens, S.; Foulkes, R.; Brown, D.; Robinson, M.; Bourne, T. Mechanism of action of certolizumab pegol (CDP870): In vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm. Bowel Dis. 2007, 13, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Bloemendaal, F.M.; Levin, A.D.; Wildenberg, M.E.; Koelink, P.J.; McRae, B.L.; Salfeld, J.; Lum, J.; van der Neut Kolfschoten, M.; Claassens, J.W.; Visser, R.; et al. Anti-tumor necrosis factor with a glyco-engineered Fc-region has increased efficacy in mice with colitis. Gastroenterology 2017, 153, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Ehrenstein, M.R. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J. Exp. Med. 2016, 213, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; Mauri, C.; Ehrenstein, M.R. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J. Exp. Med. 2007, 204, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, L.J.; Zochling, J.; Boonen, A.; Singh, J.A.; Veras, M.M.; Tanjong Ghogomu, E.; Benkhalti Jandu, M.; Tugwell, P.; Wells, G.A. TNF-alpha inhibitors for ankylosing spondylitis. Cochrane Database Syst. Rev. 2015, 3, CD005468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Jobanputra, P.; Barton, P.; Jowett, S.; Bryan, S.; Clark, W.; Fry-Smith, A.; Burls, A. A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technol. Assess. 2006, 10, 1–229. [Google Scholar] [CrossRef]
- Feagan, B.G.; McDonald, J.W.; Panaccione, R.; Enns, R.A.; Bernstein, C.N.; Ponich, T.P.; Bourdages, R.; Macintosh, D.G.; Dallaire, C.; Cohen, A.; et al. Methotrexate in combination with infliximab is no more effective than infliximab alone in patients with Crohn’s disease. Gastroenterology 2014, 146, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.; Tsynman, D.N.; Kim, A.; Arora, J.; Pietras, T.; Messing, S.; St Hilaire, L.; Yoon, S.; Decross, A.; Shah, A.; et al. Sustained improvement in health-related quality of life measures in patients with inflammatory bowel disease receiving prolonged anti-tumor necrosis factor therapy. J. Dig. Dis. 2014, 15, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Shah, N.; Deshpande, G.; Tang, D.H.; Eisenberg, D.F. Comparing biologic cost per treated patient across indications among adult us managed care patients: A retrospective cohort study. Drugs Real World Outcomes 2016, 3, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.S.; Hart, A.; De Cruz, P. Systematic review: Predicting and optimising response to anti-TNF therapy in Crohn’s disease—Algorithm for practical management. Aliment. Pharmacol. Ther. 2016, 43, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Lemann, M. Review article: Remission rates achievable by current therapies for inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 33, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Seidman, E. Predicting durable response or resistance to antitumor necrosis factor therapy in inflammatory bowel disease. Ther. Adv. Gastroenterol. 2016, 9, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Mullikin, J.C.; Facio, F.M.; Turner, C.; Cherukuri, P.F.; Blakesley, R.W.; Bouffard, G.G.; Chines, P.S.; Cruz, P.; Hansen, N.F.; et al. The ClinSeq project: Piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009, 19, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Verweij, C.L. Predicting the future of anti-tumor necrosis factor therapy. Arthritis Res. Ther. 2009, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- Deora, A.; Hegde, S.; Lee, J.; Choi, C.H.; Chang, Q.; Lee, C.; Eaton, L.; Tang, H.; Wang, D.; Lee, D.; et al. Transmembrane tnf-dependent uptake of anti-TNF antibodies. mAbs 2017, 9, 680–695. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Chowers, Y. Review article: Loss of response to anti-TNF treatments in Crohn’s disease. Aliment. Pharmacol. Ther. 2011, 33, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Noman, M.; Van Assche, G.; Baert, F.; D’Haens, G.; Rutgeerts, P. Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn’s disease. Gut 2007, 56, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Steenholdt, C.; Svenson, M.; Bendtzen, K.; Thomsen, O.O.; Brynskov, J.; Ainsworth, M.A. Acute and delayed hypersensitivity reactions to infliximab and adalimumab in a patient with Crohn’s disease. J. Crohn’s Colitis 2012, 6, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Vande Casteele, N.; Gils, A.; Singh, S.; Ohrmund, L.; Hauenstein, S.; Rutgeerts, P.; Vermeire, S. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am. J. Gastroenterol. 2013, 108, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Pouillon, L.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Noman, M.; Sabino, J.; Vande Casteele, N.; Gils, A.; Vermeire, S. Mucosal healing and long-term outcomes of patients with inflammatory bowel diseases receiving clinic-based vs. trough concentration-based dosing of infliximab. Clin. Gastroenterol. Hepatol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Casteele, N.V.; Van Stappen, T.; Van Assche, G.; Ferrante, M.; Vermeire, S.; Gils, A. Patients with high anti-drug antibody titers require a higher cumulative infliximab dose to achieve target drug concentrations: A post-hoc analysis of the taxit trial. Am. J. Gastroenterol. 2016, 111, S319. [Google Scholar]
- Vande Casteele, N.; Compernolle, G.; Ballet, V.; Van Assche, G.; Gils, A.; Vermeire, S.; Rutgeerts, P.J. Results on the optimisation phase of the prospective controlled trough level adapted infliximab treatment (taxit) trial. Gastroenterology 2012, 142, S211–S212. [Google Scholar] [CrossRef]
- Nunez Martinez, O.; Ripoll Noiseux, C.; Carneros Martin, J.A.; Gonzalez Lara, V.; Gregorio Maranon, H.G. Reactivation tuberculosis in a patient with anti-TNF-alpha treatment. Am. J. Gastroenterol. 2001, 96, 1665–1666. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, F.; Chen, J.W.; Wang, J. Risk of tuberculosis infection in anti-TNF-alpha biological therapy: From bench to bedside. J. Microbiol. Immunol. Infect. 2014, 47, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gasalla, M.; Fernandez-Baca, V.; Juan-Mas, A.; Payeras-Cifre, A.; Cifuentes-Luna, C.; Taberner-Ferrer, R.; Riera-Oliver, J.; Ros-Villamajo, I.; Navarro-Fernandez, V.; Morey Torrandell, C.; et al. Use of Quantiferon-TB-Gold in tube(®) test for detecting latent tuberculosis in patients considered as candidates for anti-TNF therapy in routine clinical practice. Enferm. Infecc. Microbiol. Clin. 2013, 31, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Fellermann, K. Adverse events of tumor necrosis factor inhibitors. Dig. Dis. 2013, 31, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Pellegrini, M.; Hall, H.; Sabbagh, L.; Ono, N.; Elford, A.R.; Mak, T.W.; Ohashi, P.S. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Investig. 2007, 117, 3833–3845. [Google Scholar] [PubMed]
- Pereira, R.; Lago, P.; Faria, R.; Torres, T. Safety of anti-TNF therapies in immune-mediated inflammatory diseases: Focus on infections and malignancy. Drug Dev. Res. 2015, 76, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L. Inflammatory bowel disease therapies and cancer risk: Where are we and where are we going? Gut 2012, 61, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Beyaert, R.; Beaugerie, L.; Van Assche, G.; Brochez, L.; Renauld, J.C.; Viguier, M.; Cocquyt, V.; Jerusalem, G.; Machiels, J.P.; Prenen, H.; et al. Cancer risk in immune-mediated inflammatory diseases (IMID). Mol. Cancer 2013, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Kaltsonoudis, E.; Voulgari, P.V.; Konitsiotis, S.; Drosos, A.A. Demyelination and other neurological adverse events after anti-TNF therapy. Autoimmun. Rev. 2014, 13, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kemanetzoglou, E.; Andreadou, E. Cns demyelination with TNF-alpha blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Genovese, M.C.; Moreland, L.W. Demyelinating and neurologic events reported in association with tumor necrosis factor alpha antagonism: By what mechanisms could tumor necrosis factor alpha antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis Rheum. 2001, 44, 1977–1983. [Google Scholar] [CrossRef]
- Wang, F.; Lin, X.; Zhao, Q.; Li, J. Adverse symptoms with anti-TNF-alpha therapy in inflammatory bowel disease: Systematic review and duration-response meta-analysis. Eur. J. Clin. Pharmacol. 2015, 71, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Druce, K.L.; Bhattacharya, Y.; Jones, G.T.; Macfarlane, G.J.; Basu, N. Most patients who reach disease remission following anti-TNF therapy continue to report fatigue: Results from the British society for rheumatology biologics register for rheumatoid arthritis. Rheumatology 2016, 55, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- Perez-De-Lis, M.; Retamozo, S.; Flores-Chavez, A.; Kostov, B.; Perez-Alvarez, R.; Brito-Zeron, P.; Ramos-Casals, M. Autoimmune diseases induced by biological agents. A review of 12,731 cases (biogeas registry). Expert Opin. Drug Saf. 2017, 16, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Tillack, C.; Ehmann, L.M.; Friedrich, M.; Laubender, R.P.; Papay, P.; Vogelsang, H.; Stallhofer, J.; Beigel, F.; Bedynek, A.; Wetzke, M.; et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-gamma-expressing Th1 cells and IL-17a/IL-22-expressing Th17 cells and respond to anti-IL-12/IL-23 antibody treatment. Gut 2014, 63, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.L.; Fraunfelder, F.W.; Rosenbaum, J.T. Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum. 2007, 56, 3248–3252. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Houvenagel, E.; Goeb, V.; Fouache, D.; Martin, A.; Le Dantec, P.; Dernis, E.; Wendling, D.; Ansemant, T.; Berthelot, J.M.; et al. Development of inflammatory bowel disease during anti-TNF-alpha therapy for inflammatory rheumatic disease: A nationwide series. Jt. Bone Spine 2012, 79, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Prati, C. Paradoxical effects of anti-TNF-alpha agents in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.L.; Gadola, S.; Edwards, C.J. Anti-TNF-induced lupus. Rheumatology 2009, 48, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Steiner, G.; Graninger, W.B.; Hofler, E.; Steiner, C.W.; Smolen, J.S. Effects of short-term infliximab therapy on autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. Dual inhibition of TNFR1 and IFNAR1 in imiquimod-induced psoriasiform skin inflammation in mice. J. Immunol. 2015, 194, 5094–5102. [Google Scholar] [CrossRef] [PubMed]
- Joyau, C.; Veyrac, G.; Dixneuf, V.; Jolliet, P. Anti-tumour necrosis factor alpha therapy and increased risk of de novo psoriasis: Is it really a paradoxical side effect? Clin. Exp. Rheumatol. 2012, 30, 700–706. [Google Scholar] [PubMed]
- Kodama, S.; Davis, M.; Faustman, D.L. The therapeutic potential of tumor necrosis factor for autoimmune disease: A mechanistically based hypothesis. Cell. Mol. Life Sci. CMLS 2005, 62, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Puimege, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, K.; de Haard, H.; Beirnaert, E.; Dreier, T.; Lauwereys, M.; Huyck, L.; Van Huysse, J.; Demetter, P.; Steidler, L.; Remaut, E.; et al. Orally administered L. lactis secreting an anti-TNF nanobody demonstrate efficacy in chronic colitis. Mucosal Immunol. 2010, 3, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ubah, O.C.; Steven, J.; Kovaleva, M.; Ferguson, L.; Barelle, C.; Porter, A.J.R.; Barelle, C.J. Novel, anti-hTNF-alpha variable new antigen receptor formats with enhanced neutralizing potency and multifunctionality, generated for therapeutic development. Front. Immunol. 2017, 8, 1780. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, Z.; Wu, F.; Tan, J.; Shen, Y.; Li, E.; Dai, J.; Shen, R.; Li, G.; Wu, J.; et al. A variant of TNFR2-fc fusion protein exhibits improved efficacy in treating experimental rheumatoid arthritis. PLoS Comput. Biol. 2010, 6, e1000669. [Google Scholar] [CrossRef] [PubMed]
- Papp, K. Clinical development of onercept, a tumor necrosis factor binding protein, in psoriasis. Curr. Med. Res. Opin. 2010, 26, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.; Sandborn, W.J.; Fedorak, R.N.; Rachmilewitz, D.; Tarabar, D.; Gibson, P.; Haagen Nielsen, O.; Wild, G.; Schreiber, S.; Pena Rossi, C.; et al. Onercept for moderate-to-severe Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 2006, 4, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Hettinghouse, A.; Liu, C. The role of progranulin in arthritis. Ann. N. Y. Acad. Sci. 2016, 1383, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.M.Z.; Mundra, J.J.; Jian, J.L.; Tian, Q.Y.; Gonzalez-Gugel, E.; Richbourgh, B.; Liu, C.J. Progranulin inhibition of TNF alpha. Immunol. Cell Biol. 2014, 92, 299–300. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Alleyne, C.H., Jr.; Dhandapani, K.M. TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci. Lett. 2013, 542, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, R.K.; Zhou, Q.H.; Hui, E.K.; Lu, J.Z.; Boado, R.J.; Pardridge, W.M. Pharmacokinetics and brain uptake of an IgG-TNF decoy receptor fusion protein following intravenous, intraperitoneal, and subcutaneous administration in mice. Mol. Pharm. 2013, 10, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gong, H.; Zhu, H.; Ji, Q.; Su, P.; Liu, P.; Cao, S.; Yao, J.; Jiang, L.; Han, M.; et al. A novel small-molecule tumor necrosis factor alpha inhibitor attenuates inflammation in a hepatitis mouse model. J. Biol. Chem. 2014, 289, 12457–12466. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mrna degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Cheng, X.; Staufenbiel, M.; Li, R.; Shen, Y. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of beta-secretase in a mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e55091. [Google Scholar]
- Alkam, T.; Nitta, A.; Mizoguchi, H.; Saito, K.; Seshima, M.; Itoh, A.; Yamada, K.; Nabeshima, T. Restraining tumor necrosis factor-alpha by thalidomide prevents the amyloid beta-induced impairment of recognition memory in mice. Behav. Brain Res. 2008, 189, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and neurodegenerative diseases. Biofactors 2013, 39, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Semmler, J.; Gebert, U.; Eisenhut, T.; Moeller, J.; Schonharting, M.M.; Allera, A.; Endres, S. Xanthine derivatives: Comparison between suppression of tumour necrosis factor-alpha production and inhibition of camp phosphodiesterase activity. Immunology 1993, 78, 520–525. [Google Scholar] [PubMed]
- Brustolim, D.; Ribeiro-dos-Santos, R.; Kast, R.E.; Altschuler, E.L.; Soares, M.B. A new chapter opens in anti-inflammatory treatments: The antidepressant bupropion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. Int. Immunopharmacol. 2006, 6, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Becnel, J.; Zerfaoui, M.; Rohatgi, R.; Boulares, A.H.; Nichols, C.D. Serotonin 5-hydroxytryptamine(2A) receptor activation suppresses tumor necrosis factor-alpha-induced inflammation with extraordinary potency. J. Pharmacol. Exp. Ther. 2008, 327, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Siegel, R.M. Wishing away inflammation? New links between serotonin and TNF signaling. Mol. Interve. 2009, 9, 299–301. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Faustman, D.L. TNF, TNF inducers, and TNFR2 agonists: A new path to type 1 diabetes treatment. Diabetes/Metab. Res. Rev. 2018, 34, e2941. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Vermeire, S. Paradoxical inflammation induced by anti-TNF agents in patients with IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Liebig, T.; Guenzi, E.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Antagonistic TNF receptor one-specific antibody (ATROSAB): Receptor binding and in vitro bioactivity. PLoS ONE 2013, 8, e72156. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Munkel, S.; Neumeyer, J.; Muller, D.; Branschadel, M.; Scheurich, P.; Pfizenmaier, K. A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J. Immunother. 2008, 31, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Abe, Y.; Ohkawa, A.; Nomura, T.; Minowa, K.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 2009, 30, 6638–6647. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, M.; Isoda, K.; Kamada, H.; Kobayashi, T.; Tsunoda, S.; Tsutsumi, Y.; Niida, T.; Kujiraoka, T.; Ishigami, N.; Ishihara, M.; et al. Novel TNF-alpha receptor 1 antagonist treatment attenuates arterial inflammation and intimal hyperplasia in mice. J. Atheroscler. Thromb. 2012, 19, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.M.; Davies, M.; Mistry, P.; Green, P.; Giddins, G.; Feldmann, M.; Stoop, A.A.; Brennan, F.M. Selective blockade of tumor necrosis factor receptor I inhibits proinflammatory cytokine and chemokine production in human rheumatoid arthritis synovial membrane cell cultures. Arthritis Rheum. 2013, 65, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Bertok, S.; Wilson, M.R.; Morley, P.J.; de Wildt, R.; Bayliffe, A.; Takata, M. Selective inhibition of intra-alveolar p55 TNF receptor attenuates ventilator-induced lung injury. Thorax 2012, 67, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Wakabayashi, K.; Bertok, S.; Oakley, C.M.; Patel, B.V.; O’Dea, K.P.; Cordy, J.C.; Morley, P.J.; Bayliffe, A.I.; Takata, M. Inhibition of TNF receptor p55 by a domain antibody attenuates the initial phase of acid-induced lung injury in mice. Front. Immunol. 2017, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.C.; Wurthner, J.U.; Morley, P.J.; Birchler, M.A.; Lambert, J.; Albayaty, M.; Serone, A.P.; Wilson, R.; Chen, Y.; Forrest, R.M.; et al. Autoantibodies to variable heavy (VH) chain ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-alpha receptor 1. J. Clin. Immunol. 2013, 33, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Cordy, J.C.; Morley, P.J.; Wright, T.J.; Birchler, M.A.; Lewis, A.P.; Emmins, R.; Chen, Y.Z.; Powley, W.M.; Bareille, P.J.; Wilson, R.; et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-alpha receptor 1. Clin. Exp. Immunol. 2015, 182, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.; Bayliffe, A.; O’Kane, C.M.; Wright, T.; Serone, A.; Bareille, P.J.; Brown, V.; Hamid, U.I.; Chen, Y.; Wilson, R.; et al. Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Chou, F.C.; Chen, S.J.; Lin, S.H.; Chang, D.M.; Sytwu, H.K. Targeting pre-ligand assembly domain of TNFR1 ameliorates autoimmune diseases—an unrevealed role in downregulation of Th17 cells. J. Autoimmun. 2011, 37, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.M.; Zheng, L.; Chan, F.K.; Lenardo, M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat. Med. 2005, 11, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Vunnam, N.; Lewis, A.K.; Chiu, T.L.; Brummel, B.E.; Schaaf, T.M.; Grant, B.D.; Bawaskar, P.; Thomas, D.D.; Sachs, J.N. An innovative high-throughput screening approach for discovery of small molecules that inhibit TNF receptors. SLAS Discov. 2017, 22, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, R.; Hu, H.; Hu, Y.; Chen, X. Modulation of regulatory T cell activity by TNF receptor type II-targeting pharmacological agents. Front. Immunol. 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Polz, J.; Remke, A.; Weber, S.; Schmidt, D.; Weber-Steffens, D.; Pietryga-Krieger, A.; Muller, N.; Ritter, U.; Mostbock, S.; Mannel, D.N. Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun. Inflamm. Dis. 2014, 2, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Zhang, H.; Wu, M.; Wang, Y. Myeloid-derived suppressor cells protect mouse models from autoimmune arthritis via controlling inflammatory response. Inflammation 2014, 37, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.; Falter, L.; Weber, S.; Muller, N.; Molitor, K.; Zeller, D.; Weber-Steffens, D.; Hehlgans, T.; Wajant, H.; Mostbock, S.; et al. Chronic inflammation increases the sensitivity of mouse Treg for TNFR2 costimulation. Front. Immunol. 2017, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, H.; Stueber, D.; Banner, D.; Mackay, F.; Lesslauer, W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J. Biol. Chem. 1993, 268, 26350–26357. [Google Scholar] [PubMed]
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016, 213, 1881–1900. [Google Scholar] [CrossRef] [PubMed]
- Lamontain, V.; Schmid, T.; Weber-Steffens, D.; Zeller, D.; Jenei-Lanzl, Z.; Wajant, H.; Straub, R.H.; Mannel, D.N. Stimulation of TNF receptor type 2 expands regulatory T cells and ameliorates established collagen-induced arthritis in mice. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Torrey, H.; Butterworth, J.; Zheng, H.; Faustman, D.L. Treg activation defect in type 1 diabetes: Correction with TNFR2 agonism. Clin. Transl. Immunol. 2016, 5, e56. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Herrera, P.L.; Harlan, D.M.; Vassalli, P. A mouse CD8 T cell-mediated acute autoimmune diabetes independent of the perforin and fas cytotoxic pathways: Possible role of membrane TNF. Proc. Natl. Acad. Sci. USA 2000, 97, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Zissimopoulos, J.M.; Barthold, D.; Brinton, R.D.; Joyce, G. Sex and race differences in the association between statin use and the incidence of Alzheimer disease. JAMA Neurol. 2017, 74, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Sabbagh, M.; Connor, D.; Soares, H.; Lopez, J.; Stankovic, G.; Johnson-Traver, S.; Ziolkowski, C.; Browne, P. Statin therapy in Alzheimer’s disease. Acta Neurol. Scand. Suppl. 2006, 185, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Nijholt, I.M.; Ostroveanu, A.; Ten Bosch, Q.; Luiten, P.G.; Eisel, U.L. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J. Alzheimer’s Dis. JAD 2008, 13, 111–122. [Google Scholar] [CrossRef]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; He, J.; Shirota, H.; Trivett, A.L.; Yang; Klinman, D.M.; Oppenheim, J.J.; Chen, X. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci. Signal. 2018, 11, eaan0790. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.L.; Wang, L.; Okubo, Y.; Burger, D.; Ban, L.; Man, G.; Zheng, H.; Schoenfeld, D.; Pompei, R.; Avruch, J.; et al. Proof-of-concept, randomized, controlled clinical trial of Bacillus-Calmette-Guerin for treatment of long-term type 1 diabetes. PLoS ONE 2012, 7, e41756. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, A.; Buzzi, M.G.; Giugni, E.; Sabatini, U.; Bastianello, S.; Pozzilli, C.; Salvetti, M.; Ristori, G. The effect of bacille calmette-guerin on the evolution of new enhancing lesions to hypointense T1 lesions in relapsing remitting MS. J. Neurol. 2003, 250, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Ristori, G.; Romano, S.; Cannoni, S.; Visconti, A.; Tinelli, E.; Mendozzi, L.; Cecconi, P.; Lanzillo, R.; Quarantelli, M.; Buttinelli, C.; et al. Effects of Bacille Calmette-Guerin after the first demyelinating event in the CNS. Neurology 2014, 82, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Secher, T.; Ezhevsky, S.A.; Janot, L.; Steed, P.M.; O’Brien, C.; Eivazi, A.; Kung, J.; Nguyen, D.H.; Doberstein, S.K.; et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J. Immunol. 2007, 179, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.L.; Di Polo, A. Neuroinflammation in glaucoma: Soluble tumor necrosis factor alpha and the connection with excitotoxic damage. Neural Regener. Res. 2016, 11, 424–426. [Google Scholar]
- Bedrosian, T.A.; Weil, Z.M.; Nelson, R.J. Chronic dim light at night provokes reversible depression-like phenotype: Possible role for TNF. Mol. Psychiatry 2013, 18, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chiu, F.L.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Chen, Y.C.; Kuo, H.C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef] [PubMed]
- Winsauer, C.; Kruglov, A.A.; Chashchina, A.A.; Drutskaya, M.S.; Nedospasov, S.A. Cellular sources of pathogenic and protective TNF and experimental strategies based on utilization of TNF humanized mice. Cytokine Growth Factor Rev. 2014, 25, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tumanov, A.V.; Grivennikov, S.I.; Kruglov, A.A.; Shebzukhov, Y.V.; Koroleva, E.P.; Piao, Y.; Cui, C.Y.; Kuprash, D.V.; Nedospasov, S.A. Cellular source and molecular form of TNF specify its distinct functions in organization of secondary lymphoid organs. Blood 2010, 116, 3456–3464. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Corazza, N.; Eichenberger, S.; Eugster, H.P.; Mueller, C. Nonlymphocyte-derived tumor necrosis factor is required for induction of colitis in recombination activating gene (RAG)2(−/−) mice upon transfer of CD4(+)CD45RB(hi) T cells. J. Exp. Med. 1999, 190, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008, 205, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Boulougouris, G.; Manoloukos, M.; Armaka, M.; Apostolaki, M.; Pizarro, T.; Kotlyarov, A.; Forster, I.; Flavell, R.; Gaestel, M.; et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J. Exp. Med. 2002, 196, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Drutskaya, M.S.; Efimov, G.A.; Kruglov, A.A.; Nedospasov, S.A. Can we design a better anti-cytokine therapy? J. Leukoc. Biol. 2017, 102, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Efimov, G.A.; Kruglov, A.A.; Khlopchatnikova, Z.V.; Rozov, F.N.; Mokhonov, V.V.; Rose-John, S.; Scheller, J.; Gordon, S.; Stacey, M.; Drutskaya, M.S.; et al. Cell-type-restricted anti-cytokine therapy: TNF inhibition from one pathogenic source. Proc. Natl. Acad. Sci. USA 2016, 113, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Nosenko, M.A.; Atretkhany, K.N.; Mokhonov, V.V.; Efimov, G.A.; Kruglov, A.A.; Tillib, S.V.; Drutskaya, M.S.; Nedospasov, S.A. VHH-based bispecific antibodies targeting cytokine production. Front. Immunol. 2017, 8, 1073. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Therapy: Cell-type-specific approach to TNF inhibition. Nat. Rev. Rheumatol. 2016, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Roulis, M.; Armaka, M.; Manoloukos, M.; Apostolaki, M.; Kollias, G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl. Acad. Sci. USA 2011, 108, 5396–5401. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Ospelt, C.; Pasparakis, M.; Kollias, G. The p55TNFR-IKK2-Ripk3 axis orchestrates arthritis by regulating death and inflammatory pathways in synovial fibroblasts. Nat. Commun. 2018, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Garcin, G.; Paul, F.; Staufenbiel, M.; Bordat, Y.; Van der Heyden, J.; Wilmes, S.; Cartron, G.; Apparailly, F.; De Koker, S.; Piehler, J.; et al. High efficiency cell-specific targeting of cytokine activity. Nat. Commun. 2014, 5, 3016. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Tumanov, A.V.; Liepinsh, D.J.; Kruglov, A.A.; Marakusha, B.I.; Shakhov, A.N.; Murakami, T.; Drutskaya, L.N.; Forster, I.; Clausen, B.E.; et al. Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils: Protective and deleterious effects. Immunity 2005, 22, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.; Romanelli, M.; Chiricozzi, A. A revolutionary therapeutic approach for psoriasis: Bispecific biological agents. Expert Opin. Investig. Drugs 2016, 25, 751–754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khatri, A.; Othman, A.A. Population pharmacokinetics of the TNF-alpha and IL-17A dual-variable domain antibody ABT-122 in healthy volunteers and subjects with psoriatic or rheumatoid arthritis: Analysis of phase 1 and 2 clinical trials. J. Clin. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Wagner, F.; Kivitz, A.J.; Mansikka, H.T.; Khan, N.; Othman, A.A.; Khatri, A.; Hong, F.; Jiang, P.; Ruzek, M.; et al. Safety, tolerability, and pharmacodynamics of ABT-122, a tumor necrosis factor- and interleukin-17-targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol. 2017, 69, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Li, R.; Chang, C. Bispecific Antibodies That Neutralize Both TNF-Alpha and IL-6: Novel Therapeutic Agent for Autoimmune Disease. U.S. Patent No. 9,416,197, 16 August 2016. [Google Scholar]
- Beidler, C.B.; Millican, R.L.; Na, S.; Seo, N. Anti-TNF/Anti-IL-23 Bispecific Antibodies. U.S. Patent No. 9,718,884, 1 August 2017. [Google Scholar]
- Kim, Y.; Yi, H.; Jung, H.; Rim, Y.A.; Park, N.; Kim, J.; Jung, S.M.; Park, S.H.; Park, Y.W.; Ju, J.H. A dual target-directed agent against interleukin-6 receptor and tumor necrosis factor alpha ameliorates experimental arthritis. Sci. Rep. 2016, 6, 20150. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Cohen, S.; Moreland, L.; Lium, D.; Robbins, S.; Newmark, R.; Bekker, P. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004, 50, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing leukemia stem cells to NF-kappab inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar] [PubMed]
- Kanakaraj, P.; Puffer, B.A.; Yao, X.T.; Kankanala, S.; Boyd, E.; Shah, R.R.; Wang, G.; Patel, D.; Krishnamurthy, R.; Kaithamana, S.; et al. Simultaneous targeting of TNF and Ang2 with a novel bispecific antibody enhances efficacy in an in vivo model of arthritis. mAbs 2012, 4, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.; Faurholm, B.; Dell’Accio, F.; Manzo, A.; Seed, M.; Eltawil, N.; Marrelli, A.; Gould, D.; Subang, C.; Al-Kashi, A.; et al. Human single-chain variable fragment that specifically targets arthritic cartilage. Arthritis Rheum. 2010, 62, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Huang, Y.; Liu, T.T.; Guo, R.; Wang, B.; Wang, X.L.; Chen, L.H.; Zhou, Y.; Ji, R.R.; Liu, T. TNF-alpha/TNFR1 signaling is required for the full expression of acute and chronic itch in mice via peripheral and central mechanisms. Neurosci. Bull. 2018, 34, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Lau, W.B.; Jiao, L.-Y.Y.; Liu, B.; Yuan, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.-L.L.; et al. Tumor necrosis factor-α and lymphotoxin-α mediate myocardial ischemic injury via TNF receptor 1, but are cardioprotective when activating TNF receptor 2. PLoS ONE 2013, 8, e60227. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.L.; Wang, M.; Crisostomo, P.R.; Abarbanell, A.M.; Herrmann, J.L.; Weil, B.R.; Meldrum, D.R. TNF receptor 2, not TNF receptor 1, enhances mesenchymal stem cell-mediated cardiac protection following acute ischemia. Shock 2010, 33, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Heusch, G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and yang in myocardial infarction? Circulation 2009, 119, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, F.; Yun, H.; Chen, L.; Muntner, P.; Levitan, E.B.; Safford, M.M.; Kent, S.T.; Osterman, M.T.; Lewis, J.D.; et al. Comparative effects of biologics on cardiovascular risk among older patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Di Genaro, M.S.; Cargnelutti, D.E.; Elicabe, J.R.; Lacoste, M.G.; Valdez, S.; Gomez, N.; de Guzman, A.M. Role of TNFRp55 in Yersinia enterocolitica O:3-induced arthritis: Triggering bacterial antigens and articular immune response. Rheumatology 2007, 46, 590–596. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, H.; Holtmann, H.; Brakebusch, C.; Avni, Y.S.; Sarov, I.; Nophar, Y.; Hadas, E.; Leitner, O.; Wallach, D. Antibodies to a soluble form of a tumor necrosis factor (TNF) receptor have TNF-like activity. J. Biol. Chem. 1990, 265, 14497–14504. [Google Scholar] [PubMed]
- Grell, M.; Scheurich, P.; Meager, A.; Pfizenmaier, K. TR60 and TR80 tumor necrosis factor (TNF)-receptors can independently mediate cytolysis. Lymphokine Cytokine Res. 1993, 12, 143–148. [Google Scholar] [PubMed]
- Hoos, A.; Anderson, J.; Boutin, M.; Dewulf, L.; Geissler, J.; Johnston, G.; Joos, A.; Metcalf, M.; Regnante, J.; Sargeant, I.; et al. Partnering with patients in the development and lifecycle of medicines: A call for action. Ther. Innov. Regul. Sci. 2015, 49, 929–939. [Google Scholar] [CrossRef] [PubMed]





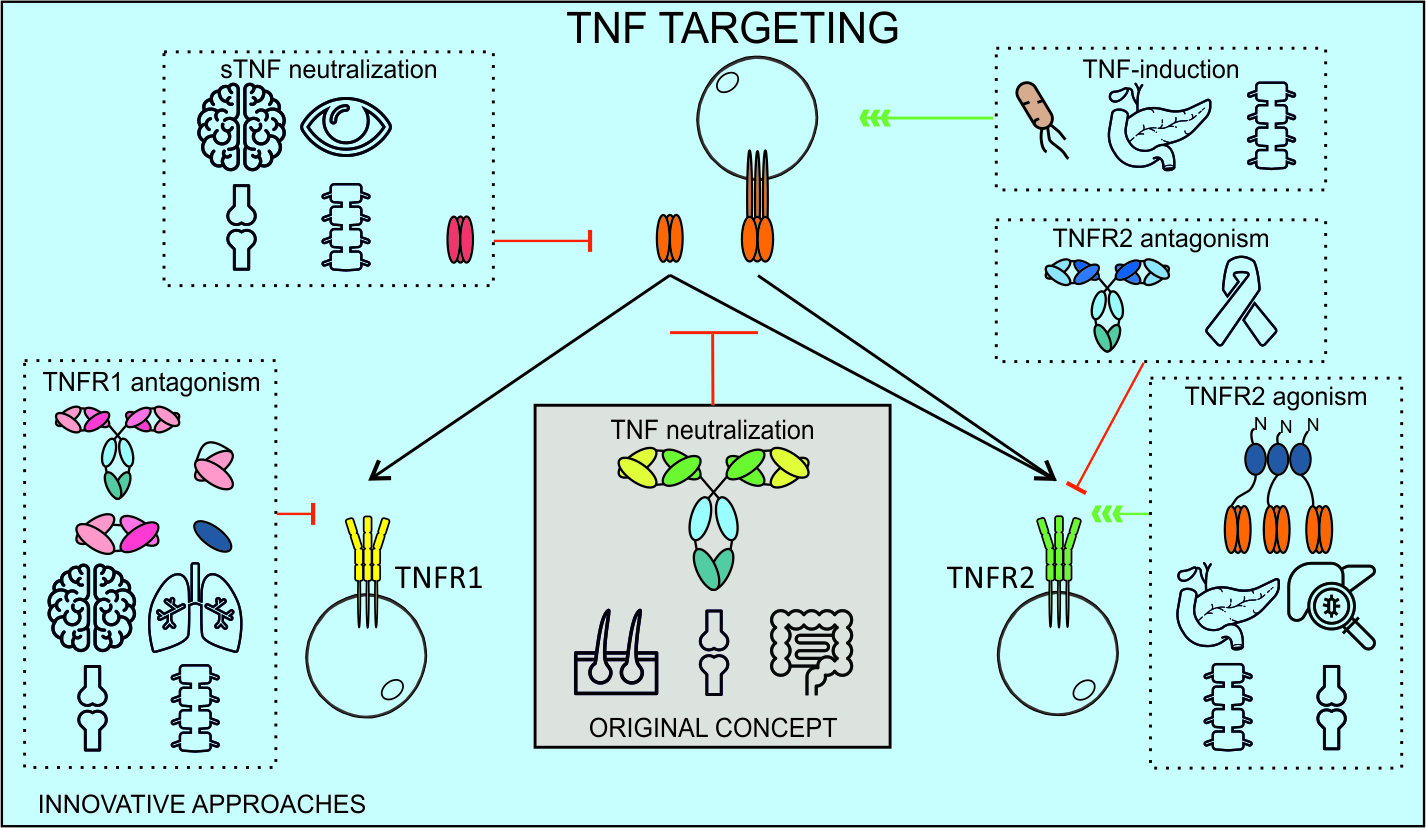{kind=link}
{kind=link}
{kind=link}
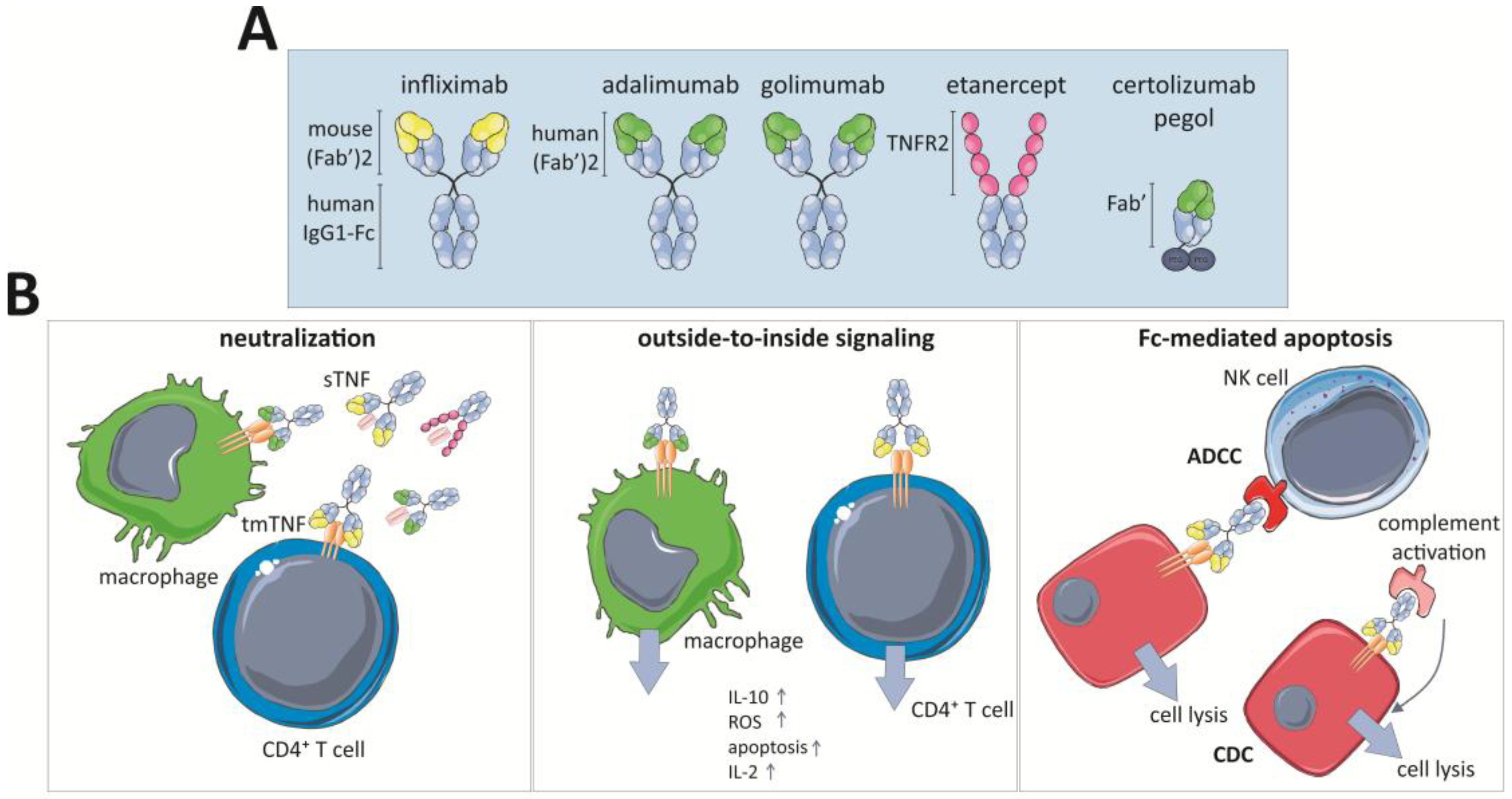{kind=link}
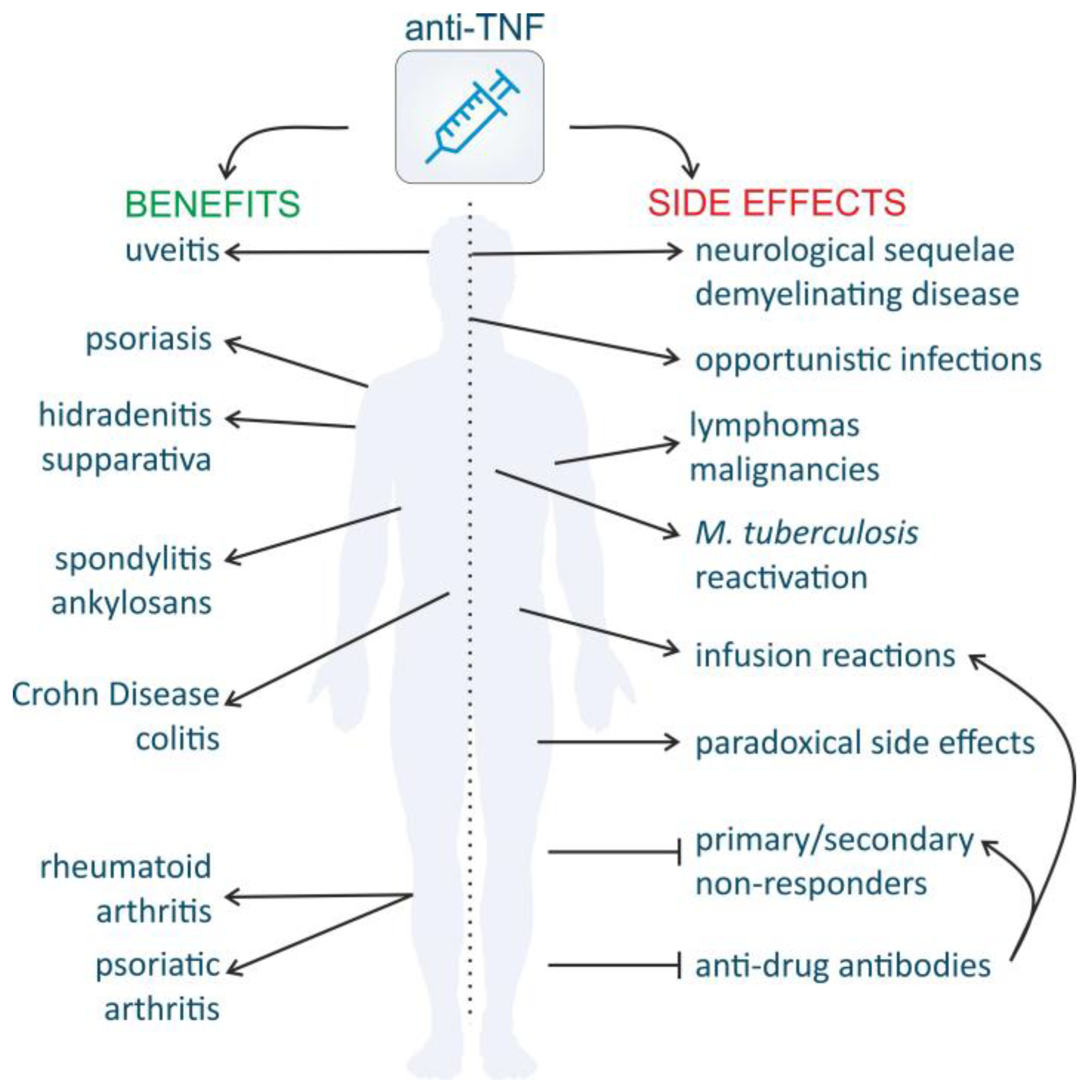{kind=link}
| Model | In Vivo/In Vitro | Mechanism | References |
|---|---|---|---|
| Multiple Sclerosis | |||
| TNFR1/TNFR2−/−, TNFR1−/−, TNFR2−/− in EAE | In vivo | TNFR1−/− mice are resistant to disease development | [160,161,171] |
| TNFR2−/− mice display exacerbated disease progression | |||
| TNF−/− and TNFR1−/− mice in EAE | In vivo | TNF protects against EAE chronicity by inducing regression of myelin-reactive T cells, independent of TNFR1 | [96] |
| TNF∆1–12 mice in EAE | In vivo | tmTNF protects against disease development | [162] |
| TNFR2−/− in EAE and bone-marrow transplantations | In vivo | TNFR2 on non-hematopoietic cells is required for Treg functioning | [181] |
| OLG-specific TNFR2 KOs (CNP-cre TNFR2fl/fl) in EAE | In vivo | OLG-TNFR2 drives OPC differentiation | [177] |
| CX3cr1-cre TNFR2fl/fl and LysM-cre TNFR2fl/fl in EAE | In vivo | TNFR2 ablation in microglia leads to early EAE onset | [173] |
| TNFR2 ablation in monocytes results in EAE suppression | |||
| TNFR1−/− that conditionally re-express TNFR1 in astrocytes in EAE (hGFAPcreT2/tnfr1cneo/cneo) | In vivo | Astrocyte TNFR1 mediates learning memory impairment | [172] |
| TNF−/−, TNFR1−/− and TNFR2−/− in CPZ model | In vivo | TNFR2 is critical for OLG proliferation and remyelination | [33] |
| TNFR2−/− in CPZ model | In vivo | Astrocyte TNFR2 mediates OPC proliferation and differentiation via CXCL12 | [179] |
| CNS-overexpressing TNF mice in TNFR1 or TNFR2 KO background | In vivo | TNFR1 induces OLG apoptosis | [157] |
| Neuron or astrocyte-overexpressing tmTNF mice | In vivo | Astrocyte tmTNF but not neuron-specific TNF triggers CNS inflammation and neurodegeneration | [145] |
| TNFR1 inhibition in EAE | In vivo | Disease development is reduced | [169,170] |
| Nanobody-based TNFR1 inhibition in EAE | In vivo | Prophylactic and therapeutic administration prevents or stops disease development | [171] |
| sTNF inhibition in EAE | In vivo | Functional outcome is improved | [165] |
| sTNF inhibition in EAE and in astrocyte-neuron coculture | In vivo | tmTNF is neuroprotective via NF-κB | [164] |
| In vitro | sTNF inhibition protects against glucose deprivation | ||
| sTNF inhibition in CPZ model | In vivo | tmTNF is neuroprotective and is needed to maintain myelin sTNF inhibits remyelination and repair | [167] |
| Astrocytes-OPC co-culture | In vitro | Astrocyte TNFR2 promotes OLG maturation via LIF | [178] |
| Rat microglia and OLG | In vitro | tmTNF kills OLGs more efficiently than sTNF | [146] |
| Alzheimer’s Disease | |||
| 5xFAD/Tg197 mice | In vivo | Peripheral hTNF mediates reduced amyloidosis and more microglial and astrocytic activation, but also synaptic loss | [185] |
| TNFR1-overexpressing primary neurons and TNFR1−/− neurons | In vitro | TNFR1 mediates Aβ-induced neuronal death | [129] |
| APP/PS1 TNFR1−/− and icv AβO injection in TNFR1−/− | In vivo | TNFR1 mediates AD-mediated choroid plexus inflammation and TNFR1+/+ mice are protected against cognitive decline, microgliosis and amyloidosis | [186] |
| APP23 TNFR1−/− | In vivo | TNFR1 signaling enhances BACE1 activity and Aβ production. Absence of TNFR1 leads to less memory deficits, neuronal loss and microglia activation | [131] |
| APP23 TNFR2−/− | In vivo | Exacerbated AD pathology | [187] |
| Nanobody-based TNFR1 inhibition icv AβO injection | In vivo | TNFR1 inhibition prevents against cognitive decline | [186] |
| sTNF inhibition in 3×Tg-AD mice | In vivo | sTNF inhibition reduces APP accumulation in hippocampus and restores synaptic dysfunction | [188,189] |
| TNFR2 inhibition in SH-SY5Y cells | In vitro | Enhances Aβ toxicity | [190] |
| Neuronal TNFR2 knockdown in 3×Tg-AD mice | In vivo | Enhances Aβ and Tau-pathology | [191] |
| Transverse hippocampus slices of WT or TNFR1−/− mice | In vitro | TNF via TNFR1 is a critical mediator of the Aβ-induced inhibition of LTP | [192] |
| Parkinson’s Disease | |||
| Double TNFR−/− mice in MPTP model | In vivo | Mice were protected against neurotoxicity, but hippocampal vulnerability increased | [193,194] |
| TNFR1−/−, TNFR2−/− mice in MPTP model | In vivo | Neither TNFR1 nor TNFR2 KO mice were protected against MPTP neurotoxicity | [195] |
| sTNF neutralization in 6-OHDA model | In vivo | Reduced nigral dopaminergic loss and microglia activation | [196,197,198] |
| Spinal Cord Injury and Traumatic Brain Injury | |||
| sTNF inhibition in SCI | In vivo | Protective | [163] |
| Double TNFR−/− mice subjected to TBI | In vivo | Bigger lesion volume and BBB impairment | [199] |
| TNFR1−/− and TNFR2−/− subjected to controlled cortical impact brain injury | In vivo | TNFR1 exacerbates cognitive functioning, TNFR2 attenuates it | [200] |
| TNFR1−/− and TNFR2−/− subjected to TBI | In vivo | TNFR1 exacerbates neurobehavioral deficits and tissue damage, TNFR2 is protective | [201] |
| Stroke, Ischemia and Oxidative Stress | |||
| Double TNFR−/− mice in stroke model | In vivo | More neuronal damage and less injury-induced microglial activation | [138] |
| TNFR1−/−, TNFR2−/− and double TNFR mice in focal cerebral ischemia/reperfusion | In vivo | TNFR1 limits neuronal damage and prevents hippocampal degeneration | [202] |
| TNFR1−/− and TNFR2−/− in model retinal ischemia | In vivo | TNFR1 augments neuronal death, TNFR2 promotes neuroprotection via PI3K-PKB/Akt pathway | [203] |
| TNFR2 agonist in LUHMES cells treated with H2O2 | In vitro | TNFR2 promotes anti-apoptotic response via PI3K-PKB/Akt pathway | [168] |
| hTNFR2-expressing OLG + TNFR2 agonist treated with H2O2 | In vitro | TNFR2 protects OPC against oxidative stress | [175] |
| Excitotoxicity and Seizures | |||
| TNFR2 agonism or TNFR1 inhibition in NMDA-induced neurodegeneration | In vivo | TNFR1 inhibition/TNFR2 agonism protects cholinergic neurons against cell death and reverts neurodegeneration-associated memory impairment | [174] |
| TNFR1−/− and TNFR2−/− mice on kainate seizures | In vivo | TNFR2 exerts anticonvulsant effects, TNFR1 mediates excitotoxicity | [204] |
| Primary cortical cells treated with glutamate | In vitro | TNFR2 protects neurons against excitotoxic insults via activation NMDA-receptor | [128] |
| Microiontophoretic administration of glutamate to TNFR1−/− or TNFR2−/− mice | In vitro | TNFR2 protects hippocampal neurons against excitotoxicity | [205] |
| Brain Inflammation | |||
| Hippocampal TNFR1−/− and TNFR2−/− neurons | In vitro | TNF vulnerability of TNFR2−/− hippocampal neurons is higher than of TNFR1−/− neurons | [130] |
| Cultured microglia | In vitro | TNFR2 upregulation after inflammatory stimuli and TNFR2-mediated induction of anti-inflammatory pathways | [182] |
| Neuropathic Pain and Hippocampal Neurogenesis | |||
| Healthy or diseased TNFR1−/− and TNFR2−/− mice | In vivo | TNFR1 is a suppressor of adult neurogenesis, absence of TNFR2 leads to reduced hippocampal neurodegeneration | [132,133,134,135] |
| TNFR1−/− and TNFR2−/− mice subjected to CCI | In vivo | TNFR1 induces a neuropathic-pain induced depression | [136] |
| Double TNFR mice and TNFR1 and TNFR2 inhibitors following CCI | In vivo | TNFR1 inhibition prolongs Wallerian degeneration and TNFR1 regulates macrophage influx | [206,207] |
| TNFR1 mediates thermal hyperalgesia | |||
| Current Approved Anti-TNF Biologics | |||||
| Drug | Biosimilars | Structure | Approved Indication | Administration Route | Ref. |
| Infliximab | CT-P13; SB2 | Chimeric mAb | RA, PA, psoriasis, AS, UC, CD, pediatric RA & CD | IV, every 8 weeks following loading at week 0, 2 and 6 | [253,254,255,256] |
| Etanercept | GP2015; SB4 | Fusion protein: human TNFR2:IgG1-Fc | RA, PA, psoriasis, AS, JIA | SC, every week or every two weeks | [256,257,258] |
| Adalimumab | ABP501; ZRC3197 | Human IgG1 mAb | RA, PA, psoriasis, AS, JIA, CD, hidradenitis suppurativa, uveitis | SC, every two weeks | [256,259] |
| Certolizumab pegol | NA | Humanized PEGylated Fab’ fragment of humanized iGG1 | RA, PA, AS, CD (only in US and Switzerland) | SC, every two weeks | [260] |
| Golimumab | NA | Human IgG1 mAb | RA, PA, AS, ulcerative colitis | SC, monthly | [261] |
| Anti-TNF Biologics in the Pipeline or Discontinued | |||||
| Drug | Biosimilar | Structure | Disease Indication | Clinical Phase/State | Ref. |
| Infliximab | BOW015; PF-06438179 | Chimeric mAb | RA | Phase III/ongoing | [256] |
| Etanercept | CHS-0214; HD203 | Fusion protein: human TNFR2:IgG1-Fc | RA, psoriasis | Phase III/ongoing | [256] |
| Adalimumab | BI695501; CHS-1420; GP-2017; M923; SB5; ZRC-3197; FKB327 | Human IgG1 mAb | RA, psoriasis, AS | Phase I, II and III/ongoing | [256] |
| Golimumab | ONS-3035 | Human IgG1 mAb | RA, PsA, AS, UC | Preclinical | |
| SSS-07 | NA | Humanized mAb | RA | Phase I | NCT02460393 |
| AVX-470; Aveximab-TNF | NA | Polyclonal bovine anti-TNF Ab | UC, NEC | Phase I | [262,263], NCT01759056 |
| CDP571 | NA | Humanized IgG4 anti-human TNF mAb | CD | Phase II/discontinued | [264] |
| Ozoralizumab | NA | Trivalent, bispecific anti-TNF Nanobody | RA, discontinued for AS, CD and PsA | Phase IIa (Japan)/ongoing | [265] |
| VHH#1-3 | NA | Bivalent Nanobody | RA | Preclinical | [266] |
| Onercept | NA | PEGylated dimeric soluble human TNFR1 | CD, psoriasis, PsA, RA, sepsis, endometriosis | Phase II and III/discontinued | [267,268] |
| Lenercept | NA | Fusion protein: soluble TNFR1:IgG1-Fc | Severe sepsis, septic shock, RA and MS | Phase II/discontinued | [53,153] |
| Hitanercept (T0001) | NA | TNFR2-Fc fusion protein | RA | Phase I (China) | [269] |
| HL036 | NA | TNFR1 fragment | Dry eye disease | Phase II | NCT03334539 |
| CytoFab | NA | Polyclonal anti-TNF Fab fragment | Severe sepsis, septic shock | Phase II/discontinued | [270] |
| Pegsunercept | NA | PEGylated sTNFR1 | RA | Phase II/discontinued | [271] |
| TNF-kinoid | NA | Vaccine to induce anti-TNF Ab, recombinant human TNF coupled to carrier protein KLH | CD, RA | Phase II/suspended | [272,273] |
| CYT007-TNFQb | NA | Vaccine to induce anti-TNF Ab, recombinant TNF coupled to virus-like particles of the bacteriophage Qbeta | Psoriasis | Phase I and II/discontinued | [274] |
| TfRMAb-TNFR | NA | Fusion protein: extracellular TNFR2 coupled to mAb against TfR | PD, AD, ischemic stroke | Preclinical | [223,242,275] |
| Disease Model | Cellular Source of TNF | Ref. | |
|---|---|---|---|
| Myeloid Cells | T Cells | ||
| T cell transfer colitis model | Pathogenic | Non-redundant | [390] |
| TNF∆ARE intestinal inflammation | Pathogenic | Pathogenic | [392] |
| TNF∆ARE joint inflammation | NA | Non-redundant | [85] |
| L. monocytogenes infection | Protective | Protective | [400] |
| M. tuberculosis infection | Dispensable 1 | Protective | [40] |
| Systemic LPS/D-Gal hepatotoxicity | Pathogenic | Dispensable | [40] |
| Autoimmune arthritis | Pathogenic | Protective | [393] |
| EAE | Pathogenic during early phase | Protective and pathogenic | [147] |
| protective in late phase | |||
| ConA-hepatitis | Pathogenic | Pathogenic | [40] |
| Diabetic nephropathy | Pathogenic | ND | [389] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. https://doi.org/10.3390/ijms19051442
Steeland S, Libert C, Vandenbroucke RE. A New Venue of TNF Targeting. International Journal of Molecular Sciences. 2018; 19(5):1442. https://doi.org/10.3390/ijms19051442
Chicago/Turabian StyleSteeland, Sophie, Claude Libert, and Roosmarijn E. Vandenbroucke. 2018. "A New Venue of TNF Targeting" International Journal of Molecular Sciences 19, no. 5: 1442. https://doi.org/10.3390/ijms19051442
APA StyleSteeland, S., Libert, C., & Vandenbroucke, R. E. (2018). A New Venue of TNF Targeting. International Journal of Molecular Sciences, 19(5), 1442. https://doi.org/10.3390/ijms19051442