The Impact of Human Papilloma Viruses, Matrix Metallo-Proteinases and HIV Protease Inhibitors on the Onset and Progression of Uterine Cervix Epithelial Tumors: A Review of Preclinical and Clinical Studies



{kind=link}
{kind=link}
Abstract
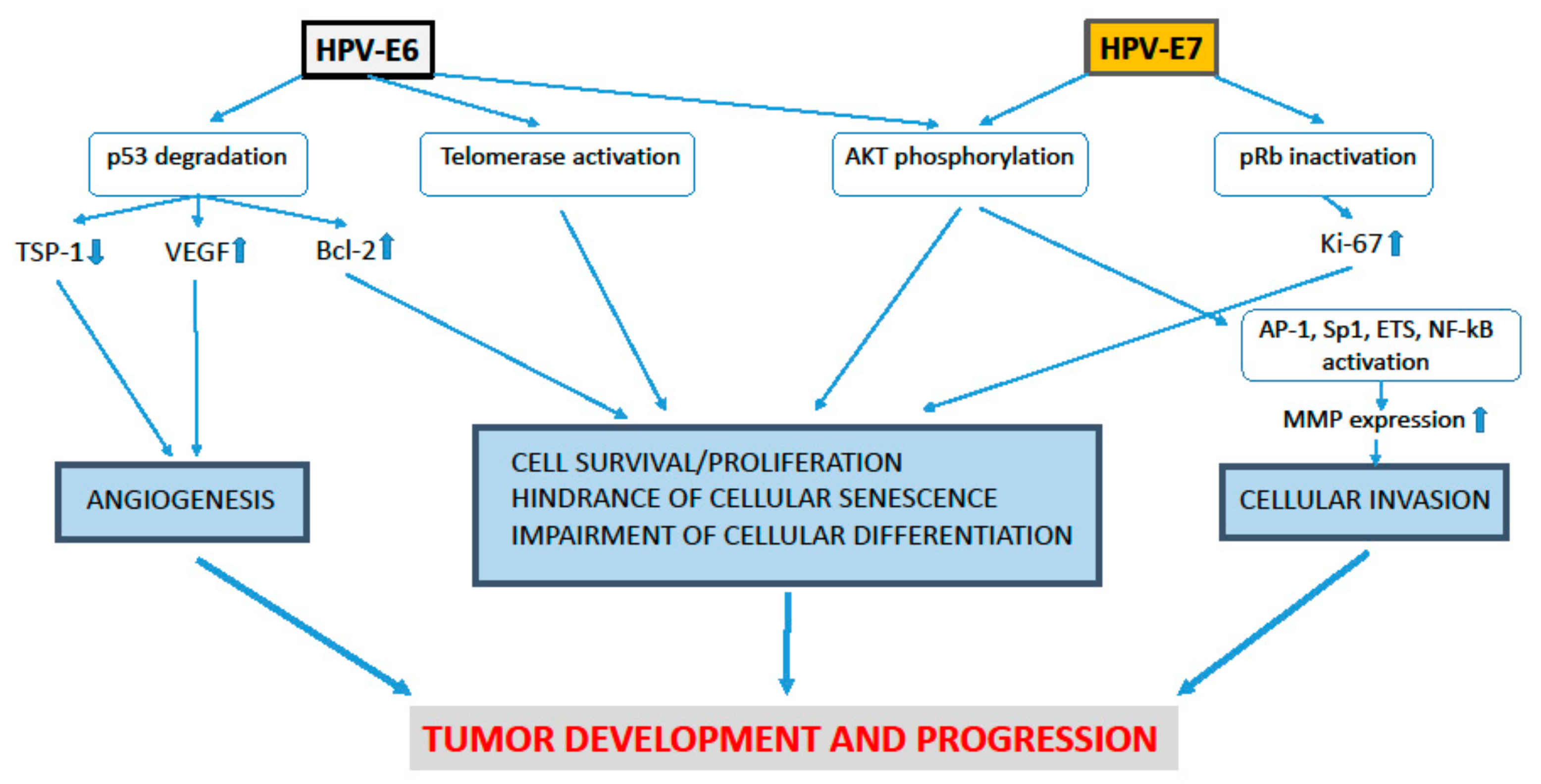
1. Role of the HPV-E5, E6 and E7 Proteins in the Development of Uterine Cervical Pre-Cancer and Cancer Lesions
2. The MMPs and Their Role in the Progression of Uterine Cervical Pre-Cancer and Cancer Lesions
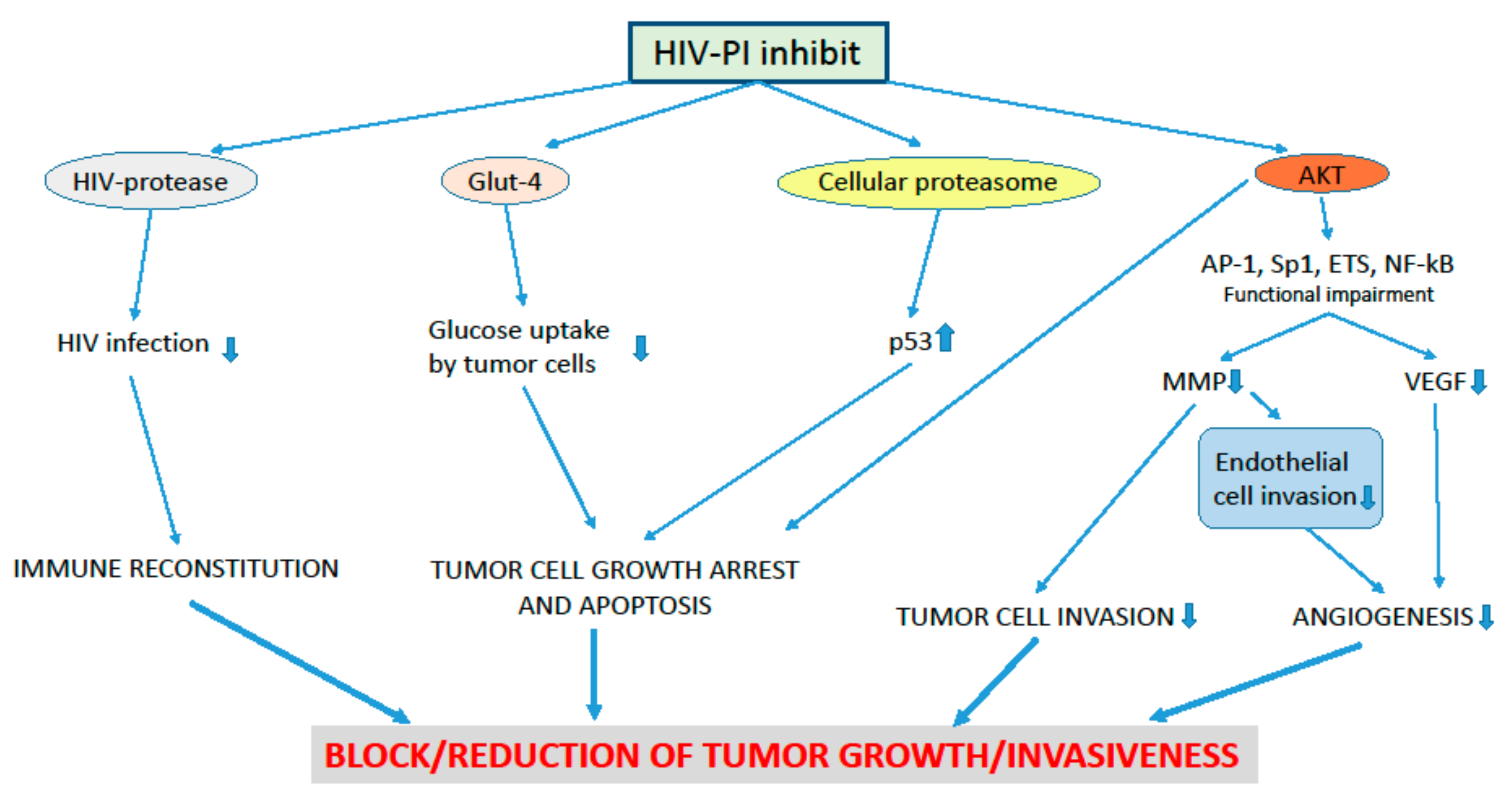
3. Effects of HIV-Protease Inhibitors on Pre-Cancer and Cancer Lesions of the Uterine Cervix
4. The Anti-Angiogenesis Effect of the HIV-PI and its Possible Impact on Pre-Cancer and Cancer Lesions of the Uterine Cervix
5. HIV-PI and HPV
6. Concluding Remarks and Future Directions
Authors Contributions
Acknowledgments
Conflicts of Interest
References
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D. Histopathology and cytopathology of cervical cancer. Dis. Markers 2007, 23, 199–212. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Laimins, L.A. Human papillomaviruses: Basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 2006, 16, 83–97. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, H.S.; Kim, S.; Oh, J.; Han, J.Y.; Lim, J.M.; Juhnn, Y.; Song, Y. Human papillomavirus type 16 E5 oncoprotein as a new target for cervical cancer treatment. Biochem. Pharmacol. 2010, 80, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Wasson, C.W.; Morgan, E.L.; Müller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Petignat, P.; Roy, M. Diagnosis and management of cervical Cancer. BMJ 2007, 335, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Basu, P.; Muwonge, R.; Banerjee, D.; Ghosh, I.; Sengupta, MM.; Das, P.; Dey, P.; Mandal, R.; Panda, C.; et al. Risk of high-grade precancerous lesions and invasive cancers in high-risk HPV-positive women with normal cervix or CIN 1 at baseline-A population-based cohort study. Int. J. Cancer 2017, 140, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, A.K.; Riemer, A.B. The invisible enemy—How human papillomaviruses avoid recognition and clearance by the host immune system. Open Virol. J. 2012, 6, 249–256. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R., Jr.; Venuti, A. hrHPV E5 oncoprotein: Immune evasion and related immunotherapies. J. Exp. Clin. Cancer Res. 2017, 36, 71–86. [Google Scholar] [CrossRef] [PubMed]
- White, A.E.; Livanos, E.M.; Tlsty, T.D. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994, 8, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.T.; Laimins, L.A. Human papillomavirus oncoproteins E6 and E7 independently abrogate the mitotic spindle checkpoint. J. Virol. 1998, 72, 1131–1137. [Google Scholar] [PubMed]
- Southern, S.A.; Noya, F.; Meyers, C.; Broker, T.R.; Chow, L.T.; Herrington, C.S. Tetrasomy is induced by human papillomavirus type 18 E7 gene expression in keratinocyte raft cultures. Cancer Res. 2001, 61, 4858–4863. [Google Scholar] [PubMed]
- Duensing, S.; Munger, K. Centrosome abnormalities and genomic instability induced by human papillomavirus oncoproteins. Prog. Cell Cycle Res. 2003, 5, 383–391. [Google Scholar] [PubMed]
- Takahashi, Y.; Ellis, L.M.; Mai, M. The angiogenic switch of human colon cancer occurs simultaneous to initiation of invasion. Oncol. Rep. 2003, 10, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Willmott, L.J.; Sumner, D.A. Antiangiogenesis agents in metastatic or recurrent cervical cancer. Gynecol. Oncol. 2010, 116, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Krill, L.S.; Krishnansu, S.T. Exploring the therapeutic rationale for angiogenesis blockade in cervical cancer. Clin. Ther. 2015, 37, 9–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saijo, Y.; Furumoto, H.; Yoshida, K.; Nishimura, M.; Irahara, M. Clinical Significance of Vascular Endothelial Growth Factor Expression and Microvessel Density in Invasive Cervical Cancer. J. Med. Investig. 2015, 62, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xiong, Y.H.; Han, J.; Guo, Z.X.; Li, Y.H.; Li, A.H.; Pei, X.Q. Contrast-enhanced ultrasonography of cervical carcinoma: Perfusion pattern and relationship with tumour angiogenesis. Br. J. Radiol. 2016, 89, 20150887. [Google Scholar] [CrossRef] [PubMed]
- Ostor, A.G. Natural history of cervical intraepithelial neoplasia: A critical review. Int. J. Gynecol. Pathol. 1993, 12, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.J.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Bosch, F.X.; Muñoz, N.; Meijer, C.J.; Shah, K.V.; Walboomers, J.M.; Herrero, R.; Franceschi, S.; International Agency for Research on Cancer; Multicentric Cervical Cancer Study Group. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: The IARC multicentric case-control study. Lancet 2002, 359, 1085–1092. [Google Scholar] [CrossRef]
- Girianelli, V.R.; Azevedo, E.; Silva, G.; Thuler, L.C. Factors associated with the risk of progression to precursor lesions or cervical cancer in women with negative cytologic findings. Int. J. Gynaecol. Obstet. 2009, 107, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Six, C.; Heard, I.; Bergeron, C.; Orth, G.; Poveda, J.D.; Zagury, P.; Cesbron, P.; Crenn-Hébert, C.; Pradinaud, R.; Sobesky, M.; et al. Comparative prevalence, incidence and short-term prognosis of cervical squamous intraepithelial lesions amongst HIV-positive and HIV-negative women. AIDS 1998, 12, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W., 3rd. Invasive and preinvasive cervical neoplasia in human immunodeficiency virus-infected women. Semin. Oncol. 2000, 27, 463–470. [Google Scholar] [PubMed]
- Sellors, J.; Lewis, K.; Kidula, N.; Muhombe, K.; Tsu, V.; Herdman, C. Screening and management of precancerous lesions to prevent cervical cancer in low-resource settings. Asian Pac. J. Cancer Prev. 2003, 4, 277–280. [Google Scholar] [PubMed]
- Omar, T.; Schwartz, S.; Hanrahan, C.; Modisenyane, T.; Tshabangu, N.; Golub, J.E.; McIntyre, J.A.; Gray, G.E.; Mohapi, L.; Martinson, N.A. Progression and regression of premalignant cervical lesions in HIV-infected women from Soweto: A prospective cohort. AIDS 2011, 25, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Blitz, S.; Baxter, J.; Raboud, J.; Walmsley, S.; Rachlis, A.; Smaill, F.; Ferenczy, A.; Coutlée, F.; Hankins, C.; Money, D. Canadian Women’s HIV Study Group: Evaluation of HIV and highly active antiretroviral therapy on the natural history of human papillomavirus infection and cervical cytopathologic findings in HIV-positive and high-risk HIV-negative women. J. Infect. Dis. 2013, 208, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Denslow, S.A.; Rositch, A.F.; Firnhaber, C.; Ting, J.; Smith, J.S. Incidence and progression of cervical lesions in women with HIV: A systematic global review. Int. J. STD AIDS 2014, 25, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Obiri-Yeboah, D.; Akakpo, P.K.; Mutocheluh, M.; Adjei-Danso, E.; Allornuvor, G.; Amoako-Sakyi, D.; Adu-Sarkodie, Y.; Mayaud, P. Epidemiology of cervical human papillomavirus (HPV) infection and squamous intraepithelial lesions (SIL) among a cohort of HIV-infected and uninfected Ghanaian women. BMC Cancer 2017, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Nappi, L.; Carriero, C.; Bettocchi, S.; Herrero, J.; Vimercati, A.; Putignano, G. Cervical squamous intraepithelial lesions of low-grade in HIV-infected women: Recurrence, persistence and progression, in treated and untreated women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2005, 121, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Ntekim, A.; Campbell, O.; Rothenbacher, D. Optimal management of cervical cancer in HIV-positive patients: A systematic review. Cancer Med. 2015, 4, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.M. AIDS-related malignancies: The emerging epidemic. J. Natl. Cancer Inst. 1993, 85, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Cu-Uvin, S. Association of HIV viral load and CD4 cell count with human papillomavirus detection and clearance in HIV-infected women initiating highly active antiretroviral therapy. HIV Med. 2012, 13, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Buonaguro, F.M.; Beth-Giraldo, E.; Giraldo, G. Human immunodeficiency virus type 1 tat gene enhances human papillomavirus early gene expression. Intervirology 1993, 36, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Nyagol, J.; Leucci, E.; Onnis, A.; De Falco, G.; Tigli, C.; Sanseverino, F.; Torriccelli, M.; Palummo, N.; Pacenti, L.; Santopietro, R.; et al. The effects of HIV-1 Tat protein on cell cycle during cervical carcinogenesis. Cancer Biol. Ther. 2006, 5, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Yochim, J.M.; Kang, M.K.; Shin, K.H.; Christensen, R.; Park, N.H. HIV-1 Tat enhances replicative potential of human oral keratinocytes harboring HPV-16 genome. Int. J. Oncol. 2008, 33, 777–782. [Google Scholar] [PubMed]
- Barillari, G.; Palladino, C.; Bacigalupo, I.; Leone, P.; Falchi, M.; Ensoli, B. Entrance of the Tat protein of HIV-1 into human uterine cervical carcinoma cells causes up-regulation of HPV-E6 expression and a decrease in p53 protein levels. Oncol. Lett. 2016, 12, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Barillari, G.; Ensoli, B. Angiogenic effects of extracellular HIV-1 Tat protein and its role in the pathogenesis of AIDS-associated Kaposi’s sarcoma. Clin. Microbiol. Rev. 2002, 15, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Buonaguro, L.; Giorgi-Rossi, P.; Buonaguro, F.M. Viral and cellular Biomarkers in the diagnosis of cervical intraepithelial neoplasia and cancer. Biomed. Res. Int. 2013, 2013, 519619. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Zhu, Y.; Yang, L.; Zhang, X.; Liu, L.; Ren, C. Diagnostic performance of HPV E6/E7 mRNA assay for detection of cervical high-grade intraepithelial neoplasia and cancer among women with ASCUS Papanicolaou smears. Arch. Gynecol. Obstet. 2017, 297, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.J.; Liu, H.; Wu, D.; Tang, Z.H.; Shen, Y.C.; Guo, L. E6/E7 proteins are potential markers for the screening and diagnosis of cervical pre-cancerous lesions and cervical cancer in a Chinese population. Oncol. Lett. 2017, 14, 6251–6258. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.W.; Lin, M.C.; Hsiao, C.H.; Lin, Y.T.; Kuo, K.T. Papillary squamous intraepithelial lesions of the uterine cervix: Human papillomavirus-dependent changes in cell cycle expression and cytologic features. Hum. Pathol. 2010, 41, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Bucchi, L.; Cortecchia, S.; Galanti, G.; Sgadari, C.; Costa, S.; De Lillo, M.; Caparra, L.; Barillari, G.; Monini, P.; Nannini, R.; et al. Follow-up study of patients with cervical intraepithelial neoplasia grade 1 overexpressing p16ink4a. Int. J. Gynecol. Cancer 2013, 23, 1663–1669. [Google Scholar] [CrossRef]
- Calil, L.N.; Edelweiss, M.I.; Meurer, L.; Igansi, C.N.; Bozzetti, M.C. p16 INK4a and Ki67 expression in normal, dysplastic and neoplastic uterine cervical epithelium and human papillomavirus (HPV) infection. Pathol. Res. Pract. 2014, 210, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Torres, A.; López-Urrutia, E.; García-Castillo, V.; Jacobo-Herrera, N.; Herrera, L.A.; Peralta-Zaragoza, O.; López-Camarillo, C.; Cantú De Leon, D.; Fernández-Retana, J.; Cerna-Cortés, J.F.; et al. MicroRNAs in cervical cancer: Evidences for a miRNA profile deregulated by HPV and its impact on radio-resistance. Molecules 2014, 19, 6263–6281. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Löffek, S.; Schilling, O.; Franzke, C.W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.G.; Parks, W.C. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015, 44–46, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Nair, S. Proteases in cardiometabolic diseases: Pathophysiology, molecular mechanisms and clinical applications. Biochem. Biophys. Acta 2015, 1852, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Irigoyen, O.; Carotti, S.; Latasa, M.U.; Uriarte, I.; Fernández-Barrena, M.G.; Elizalde, M.; Urtasun, R.; Vespasiani-Gentilucci, U.; Morini, S.; Banales, J.M.; et al. Matrix metalloproteinase-10 expression is induced during hepatic injury and plays a fundamental role in liver tissue repair. Liver Int. 2014, 34, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Ahmad, R.; Kaur, G. Matrix metalloproteinases and cancer—Roles in threat and therapy. Asian Pac. J. Cancer Prev. 2014, 15, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Ågren, M.S.; Schnabel, R.; Christensen, L.H.; Mirastschijski, U. Tumor necrosis factor-α-accelerated degradation of type I collagen in human skin is associated with elevated matrix metalloproteinase (MMP)-1 and MMP-3 ex vivo. Eur. J. Cell Biol. 2015, 94, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Xin, S.; Wang, H.; Ma, J.; Zhang, H.; Wei, H. Baicalein inhibits MMP-2 expression in human ovarian cancer cells by suppressing the p38MAPK-dependent NF-κB signaling pathway. Anticancer Drugs 2015, 26, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.C.; Jiang, Q.; Yu, Y.; Mei, J.P.; Cui, Y.K.; Zhao, W.J. Quercetin promotes cell apoptosis and inhibits the expression of MMP-9 and fibronectin via the AKT and ERK signalling pathways in human glioma cells. Neurochem. Int. 2015, 80, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.; Gotoh, K.; Tsuchiya, M.; Sakisaka, Y.; Shimauchi, H. Extracellular ATP inhibits IL-1-induced MMP-1 expression through the action of CD39/nucleotidase triphosphate dephosphorylase-1 on human gingival fibroblasts. Int. Immunopharmacol. 2013, 17, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Gilet, A.; Zou, F.; Boumenir, M.; Frippiat, J.P.; Thornton, S.N.; Lacolley, P.; Ropars, A. Aldosterone up-regulates MMP-9 and MMP-9/NGAL expression in human neutrophils through p38, ERK1/2 and PI3K pathways. Exp. Cell Res. 2015, 331, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.; Kirkpatrick, G.; Evans, M.; Gorski, G.; Foster, S.; Borghaei, R.C. IL-4 inhibition of IL-1 induced Matrix metalloproteinase-3 (MMP-3) expression in human fibroblasts involves decreased AP-1 activation via negative crosstalk involving of Jun N-terminal kinase (JNK). Exp. Cell Res. 2013, 319, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Peña, E.; de la Torre, R.; Arderiu, G.; Slevin, M.; Badimon, L. mCRP triggers angiogenesis by inducing F3 transcription and TF signalling in microvascular endothelial cells. Tromb Haemost 2017, 117, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, Y.S.; Wang, K.; Chien, H.W.; Hsieh, Y.H.; Yang, S.F. Fisetin inhibits epidermal growth factor-induced migration of ARPE-19 cells by suppression of AKT activation and Sp1-dependent MMP-9 expression. Mol. Vis. 2017, 23, 900–910. [Google Scholar] [PubMed]
- Adiseshaiah, P.; Vaz, M.; Machireddy, N.; Kalvakolanu, D.V.; Reddy, S.P. A. Fra-1-dependent, matrix metalloproteinase driven EGFR activation promotes human lung epithelial cell motility and invasion. J. Cell. Physiol. 2008, 216, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.J.; Zhao, J.; Jiang, M.L.; Zhang, L.C. ING5 inhibits cell proliferation and invasion in esophageal squamous cell carcinoma through regulation of the Akt/NF-κB/MMP-9 signaling pathway. Biochem. Biophys. Res. Commun. 2018, 496, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology 2013, 28, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; Pendas, A.M.; Sanchez, L.M.; Lopez-Otin, C. Matrix metalloproteinases in cancer: From new functions to improved inhibition strategies. Int. J. Dev. Biol. 2004, 48, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Hanzawa, M.; Shindoh, M.; Higashino, F.; Yasuda, M.; Inoue, N.; Hida, K.; Ono, M.; Kohgo, T.; Nakamura, M.; Notani, K.; et al. Hepatocyte growth factor up-regulates E1AF that induces oral squamous cell carcinoma cell invasion by activating matrix metalloproteinase genes. Carcinogenesis 2000, 21, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Jacob-Ferreira, A.L.; Schulz, R. Activation of intracellular matrix metalloproteinase-2 by reactive oxygen-nitrogen species: Consequences and therapeutic strategies in the heart. Arch. Biochem. Biophys. 2013, 540, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.L.; Liu, X.; Gao, S.Y.; Feng, H.; Jiang, Y.P.; Wang, S.S.; Yang, J.; Jiang, J.; Ma, X.R.; Tang, Y.J.; et al. WIP1 stimulates migration and invasion of salivary adenoid cystic carcinoma by inducing MMP-9 and VEGF-C. Oncotarget 2015, 6, 9031–9044. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Dier, U.; Melendez, J.A.; Hempel, N. Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim. Biophys. Acta 2015, 1852, 2593–2602. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Li, J.; Chen, X.; Chen, K.; Li, W.; Li, X.; Zhang, L.; Duan, W.; Lei, J.; Xu, Q.; et al. Lipoxin A4 Attenuates Cell Invasion by Inhibiting ROS/ERK/MMP Pathway in Pancreatic Cancer. Oxid. Med. Cell. Longev. 2016, 6815727. [Google Scholar] [CrossRef]
- Fullár, A.; Kovalszky, I.; Bitsche, M.; Romani, A.; Schartinger, V.H.; Sprinzl, G.M.; Riechelmann, H.; Dudás, J. Tumor cell and carcinoma-associated fibroblast interaction regulates matrix metalloproteinases ant their inhibitors in oral squamous cell carcinoma. Exp. Cell Res. 2012, 318, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wu, Y.; Xie, X.D.; Chu, Y.F.; Li, J.Q.; Zheng, L. c-Met identifies a population of matrix metalloproteinase 9-producing monocytes in peritumoural stroma of hepatocellular carcinoma. J. Pathol. 2015, 237, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mashhadiabbas, F.; Mahjour, F.; Mahjour, S.B.; Fereidooni, F.; Hosseini, F.S. The immunohistochemical characterization of MMP-2, MMP-10, TIMP-1, TIMP-2 and podoplanin in oral squamous cell carcinoma. Oral Surg. Oral Med. Pathol Oral. Radiol. 2012, 114, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Hui, P.; Xu, X.; Xu, L.; Hui, G.; Wu, S.; Lan, Q. Expression of MMP14 in invasive pituitary adenomas: Relationship to invasion and angiogenesis. Int. J. Clin. Exp. Pathol. 2015, 8, 3556–3567. [Google Scholar] [PubMed]
- Herszényi, L.; Barabás, L.; Hritz, I.; István, G.; Tulassay, Z. Impact of proteolytic enzymes in colorectal cancer development and progression. World J. Gastroenterol. 2014, 20, 13246–13257. [Google Scholar] [CrossRef] [PubMed]
- Figueira, R.C.; Gomes, L.R.; Neto, J.S.; Silva, F.C.; Silva, I.D.; Sogayar, M.C. Correlation between MMPs and their inhibitors in breast cancer tumor tissue specimens and in cell lines with different metastatic potential. BMC Cancer 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, D.; Zhang, W.; Zhou, J.; Tang, B.; Li, L. Matrix metalloproteinase-9 expression correlates with prognosis and involved in ovarian cancer cell invasion. Arch. Gynecol. Obstet. 2012, 286, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.H.; Tretiakova, M.S.; Liu, W.H.; Gong, C.; Farris, P.D.; Hart, J. Association of E-cadherin, matrix metalloproteinases with the progression and metastasis of hepatocellular carcinoma. Mod. Pathol. 2006, 19, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, G.; Pavlakis, K.; Parasi, A.; Kavatzas, N.; Tiniakos, D.; Karakosta, A.; Tzanakis, N.; Peros, G. The expression of matrix metalloproteinase-2 and -9 and their tissue inhibitor 2 in pancreatic ductal and ampullary carcinoma and their relation to angiogenesis and clinicopathological parameters. Anticancer Res. 2008, 28, 1875–1881. [Google Scholar] [PubMed]
- Groblewska, M.; Siewko, M.; Mroczko, B.; Szmitkowski, M. The role of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophageal cancer. Folia Histochem. Cytobiol. 2012, 50, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, S.; Mahmoudian, R.A.; Abbaszadegan, M.R.; Chamani, J.; Memar, B.; Gholamin, M. Expression analysis of matrix metalloproteinase-13 in human gastric cancer in the presence of Helicobacter Pylori infection. Cancer Biomark. 2017, 18, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, J.P.; Vornanen, J.; Tervahartiala, T.; Sorsa, T.; Bloigu, R.; Salo, T.; Tuomisto, A.; Mäkinen, M.J. Serum MMP-8 levels increase in colorectal cancer and correlate with disease course and inflammatory properties of primary tumors. Int. J. Cancer 2012, 131, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Chen, G.W.; Liu, Y.C.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Zhu, J.; Gao, H.Q.; Yin, J.; Wang, W.; et al. Secreted protein acidic and rich in cysteine (SPARC) suppresses angiogenesis by down-regulating the expression of VEGF and MMP-7 in gastric cancer. PLoS ONE 2012, 7, e44618. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behavior in vascular formation and remodeling. Mediat. Inflamm. 2013, e928315. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wen, G.; Zhang, L.; Lin, L.; Moore, A.; Wu, S.; Ye, S.; Xiao, Q. An important role of matrix metalloproteinase-8 in angiogenesis in vitro and in vivo. Cardiovasc. Res. 2013, 99, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Ke, G.; Wang, L.; Gu, Q.; Zhou, E.; He, Q.; Wang, S. Silencing Matrix Metalloproteinases 9 and 2 Inhibits Human Retinal Microvascular Endothelial Cell Invasion and Migration. Ophthalmic Res. 2015, 55, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Sharma, V. Tumour Angiogenesis and Angiogenic Inhibitors: A Review. Clin. Diagn. Res. 2015, 9, XE01–XE05. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ni, S.; Cao, Y.; Zhang, T.; Wu, T.; Yin, X.; Lang, Y.; Lu, H. The Angiogenic Effect of microRNA-21 Targeting TIMP3 through the Regulation of MMP2 and MMP9. PLoS ONE 2016, 11, e0149537. [Google Scholar] [CrossRef]
- Fukuda, H.; Mochizuki, S.; Abe, H.; Okano, H.J.; Hara-Miyauchi, C.; Okano, H.; Yamaguchi, N.; Nakayama, M.; D’Armiento, J.; Okada, Y. Host-derived MMP-13 exhibits a protective role in lung metastasis of melanoma cells by local endostatin production. Br. J. Cancer 2011, 105, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Korpi, J.T.; Kervinen, V.; Mäklin, H.; Väänänen, A.; Lahtinen, M.; Läärä, E.; Ristimäki, A.; Thomas, G.; Ylipalosaari, M.; Aström, P.; et al. Collagenase-2 (matrix metalloproteinase-8) plays a protective role in tongue cancer. Br. J. Cancer 2008, 98, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Kleifeld, O. Tumour microenvironment—Opinion: Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Abdul Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Haorah, J. The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Huang, L.; Li, J.; Qu, S.; Pan, F. Matrix metalloproteinase-14 expression and its prognostic value in cervical carcinoma. Cell Biochem. Biophys. 2014, 70, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Hotary, K.B.; Nan, B.; Bosch, F.X.; Muñoz, N.; Weiss, S.J.; Cho, K.R. Expression of membrane type 1 matrix metalloproteinase is associated with cervical carcinoma progression and invasion. Cancer Res. 2005, 65, 6543–6550. [Google Scholar] [CrossRef] [PubMed]
- Sheu, B.C.; Lien, H.C.; Ho, H.N.; Lin, H.H.; Chow, S.N.; Huang, S.C.; Hsu, S.M. Increased expression and activation of gelatinolytic matrix metalloproteinases is associated with the progression and recurrence of human cervical cancer. Cancer Res. 2003, 63, 6537–6542. [Google Scholar] [PubMed]
- Matheus, E.R.; Zonta, M.A.; Discacciati, M.G.; Paruci, P.; Velame, F.; Cardeal, L.B.; Barros, S.B.; Pignatari, A.C.; Maria-Engler, S.S. MMP-9 expression increases according to the grade of squamous intraepithelial lesion in cervical smears. Diagn. Cytopathol. 2014, 42, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Nasr, M.; Ayyad, S.B.; El-Lamie, I.K.; Mikhail, M.Y. Expression of matrix metalloproteinase-2 in preinvasive and invasive carcinoma of the uterine cervix. Gynaecol. Oncol. 2005, 26, 199–202. [Google Scholar]
- Davidson, B.; Goldberg, I.; Kopolovic, J.; Lerner-Geva, L.; Gotlieb, W.H.; Weis, B.; Ben-Baruch, G.; Reich, R. Expression of matrix metalloproteinase-9 in squamous cell carcinoma of the uterine cervix-clinicopathologic study using immunohistochemistry and mRNA in situ hybridization. Gynecol. Oncol. 1999, 72, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Branca, M.; Ciotti, M.; Giorgi, C.; Santini, D.; Di Bonito, L.; Costa, S.; Benedetto, A.; Bonifacio, D.; Di Bonito, P.; Paba, P.; et al. HPV-Pathogen ISS Study Group. Matrix metalloproteinase-2 (MMP-2) and its tissue inhibitor (TIMP-2) are prognostic factors in cervical cancer, related to invasive disease but not to high-risk human papillomavirus (HPV) or virus persistence after treatment of CIN. Anticancer Res. 2006, 26, 1543–1556. [Google Scholar] [PubMed]
- Talvensaari-Mattila, A.; Turpeenniemi-Hujanen, T. Matrix metalloproteinase 9 in the uterine cervix during tumor progression. Int. J. Gynaecol. Obstet. 2006, 92, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Van Trappen, P.O.; Ryan, A.; Carroll, M.; Lecoeur, C.; Goff, L.; Gyselman, V.G.; Young, B.D.; Lowe, D.G.; Pepper, M.S.; Shepherd, J.H.; et al. A model for co-expression pattern analysis of genes implicated in angiogenesis and tumor cell invasion in cervical cancer. Br. J. Cancer 2002, 87, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Asha Nair, S.; Karunagaran, D.; Nair, M.B.; Sudhakaran, P.R. Changes in matrix metalloproteinases and their endogenous inhibitors during tumor progression in the uterine cervix. J. Cancer Res. Clin. Oncol. 2003, 129, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Gaiotto, M.A.; Focchi, J.; Ribalta, J.L.; Stávale, J.N.; Baracat, E.C.; Lima, G.R.; Guerreiro da Silva, I.D. Comparative study of MMP-2 (matrix metalloproteinase 2) immune expression in normal uterine cervix, intraepithelial neoplasias and squamous cells cervical carcinoma. Am. J. Obstet. Gynecol. 2004, 190, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Chen, H.; Zheng, X.M.; Chen, M.L. Expression and clinical significance of high risk human papillomavirus and invasive gene in cervical carcinoma. Asian Pac. J. Trop. Med. 2017, 10, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.; de Angelo-Andrade, L.A.; Morais, S.S.; Pinto, G.A.; Chagas, C.A.; Maria-Engler, S.S.; Zeferino, L.C. Stromal cells play a role in cervical cancer progression mediated by MMP-2 protein. Eur. J. Gynaecol. Oncol. 2008, 29, 341–344. [Google Scholar] [PubMed]
- Yang, S.F.; Wang, P.H.; Lin, L.Y.; Ko, J.L.; Chen, G.D.; Yang, J.S.; Lee, H.S.; Hsieh, Y.S. A significant elevation of plasma level of matrix metalloproteinase-9 in patients with high-grade intraepithelial neoplasia and early squamous cell carcinoma of the uterine cervix. Reprod. Sci. 2007, 14, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Smola-Hess, S.; Pahne, J.; Mauch, C.; Zigrino, P.; Smola, H.; Pfister, H.J. Expression of membrane type 1 matrix metalloproteinase in papillomavirus-positive cells: Role of the human papillomavirus (HPV) 16 and HPV8 E7 gene products. J. Gen. Virol. 2005, 86, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Cardeal, L.B.; Brohem, C.A.; Correa, T.C.; Winnischofer, S.M.; Nakano, F.; Boccardo, E.; Villa, L.L.; Sogayar, M.C.; Maria-Engler, S.S. Higher expression and activity of metalloproteinases in human cervical carcinoma cell lines is associated with HPV presence. Biochem. Cell Biol. 2006, 84, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kajitani, N.; Satsuka, A.; Nakamura, H.; Sakai, H. Ras modifies proliferation and invasiveness of cells expressing human papillomavirus oncoproteins. J. Virol. 2008, 82, 8820–8827. [Google Scholar] [CrossRef] [PubMed]
- Barbaresi, S.; Cortese, M.S.; Quinn, J.; Ashrafi, G.H.; Graham, S.V.; Campo, M.S. Effects of human papillomavirus type 16 E5 deletion mutants on epithelial morphology: Functional characterization of each trans-membrane domain. J. Gen. Virol. 2010, 91, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Cardeal, L.B.; Boccardo, E.; Termini, L.; Rabachini, T.; Andreoli, M.A.; di Loreto, C.; Longatto Filho, A.; Villa, L.L.; Maria-Engler, S.S. HPV16 oncoproteins induce MMPs/RECK-TIMP-2 imbalance in primary keratinocytes: Possible implications in cervical carcinogenesis. PLoS ONE 2012, 7, e33585. [Google Scholar] [CrossRef] [PubMed]
- Kaewprag, J.; Umnajvijit, W.; Ngamkham, J.; Ponglikitmongkol, M. HPV16 oncoproteins promote cervical cancer invasiveness by upregulating specific matrix metalloproteinases. PLoS ONE 2013, 8, e71611. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Ye, M.; Zhang, W. E6/E7 oncoproteins of high risk HPV-16 upregulate MT1-MMP, MMP-2 and MMP-9 and promote the migration of cervical cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4981–4989. [Google Scholar] [PubMed]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signalling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Lee, J.J.; Komaki, R.; Herbst, R.S.; Feng, L.; Evans, W.K.; Choy, H.; Desjardins, P.; Esparaz, B.T.; Truong, M.T.; et al. Chemoradiotherapy with or without AE-941 in stage III non-small cell lung cancer: A randomized phase III trial. J. Natl. Cancer Inst. 2010, 102, 859–865. [Google Scholar] [CrossRef] [PubMed]
- El-Rayes, B.F.; Philip, P.A.; Sarkar, F.H.; Shields, A.F.; Ferris, A.M.; Hess, K.; Kaseb, A.O.; Javle, M.M.; Varadhachary, G.R.; Wolff, R.A.; et al. A phase II study of isoflavones, erlotinib and gemcitabine in advanced pancreatic cancer. Investig. New Drugs 2011, 29, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lee, H.M.; Roemer, E.J.; Musacchia, L.; Golub, L.M.; Simon, S.R. Inhibition of tumor cell invasiveness by chemically modified tetracyclines. Curr. Med. Chem. 2001, 8, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.J.; Hsu, S.F.; Li, T.M.; Hsu, H.C.; Lin, J.G.; Hsu, C.J.; Chou, M.C.; Lee, M.C.; Yang, S.F.; Fong, Y.C. Alendronate inhibits cell invasion and MMP-2 secretion in human chondrosarcoma cell line. Acta Pharmacol. Sin. 2007, 28, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R. Prinomastat, a hydroxamate-based matrix metalloproteinase inhibitor. A novel pharmacological approach for tissue remodeling-related diseases. Expert Opin. Investig. Drugs 2000, 9, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Box, G.M.; Court, W.J.; Bone, E.A.; Thomas, W.; Brown, P.D. Control of lymphatic and hematogenous metastasis of a rat mammary carcinoma by the matrix metalloproteinase inhibitor batimastat (BB-94). Cancer Res. 1996, 56, 2815–2822. [Google Scholar] [PubMed]
- Price, A.; Shi, Q.; Morris, D.; Wilcox, M.E.; Brasher, P.M.; Rewcastle, N.B.; Shalinsky, D.; Zou, H.; Appelt, K.; Johnston, R.N.; et al. Marked inhibition of tumor growth in a malignant glioma tumor model by a novel synthetic matrix metalloproteinase inhibitor AG3340. Clin. Cancer Res. 1999, 5, 845–854. [Google Scholar] [PubMed]
- Shevrin, D.H.; Gorny, K.I.; Rosol, T.J.; Kukreja, S.C. Effect of etidronate disodium on the development of bone lesions in an animal model of bone metastasis using the human prostate cancer cell line PC-3. Prostate 1991, 19, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.H.; Lee, J.; Orr, F.W. The effect of Neovastat (AE-941) on an experimental metastatic bone tumor model. Int. J. Oncol. 2002, 20, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Yang, K.W.; Hsu, Y.T.; Chang, C.L.; Yang, Y.C. The differential inhibitory effects of genistein on the growth of cervical cancer cells in vitro. Neoplasma 2001, 48, 227–233. [Google Scholar] [PubMed]
- Iwasaki, M.; Nishikawa, A.; Fujimoto, T.; Akutagawa, N.; Manase, K.; Endo, T.; Yoshida, K.; Maekawa, R.; Yoshioka, T.; Kudo, R. Anti-invasive Effect of MMI-166, a New Selective Matrix Metalloproteinase Inhibitor, in Cervical Carcinoma Cell Lines. Gynecol. Oncol. 2002, 85, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bramhall, S.R.; Schulz, J.; Nemunaitis, J.; Brown, P.D.; Baillet, M.; Buckels, J.A. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 2002, 87, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Pavlaki, M.; Zucker, S. Matrix metalloproteinase inhibitors (MMPIs): The beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 2003, 22, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Bissett, D.; O’Byrne, K.J.; von Pawel, J.; Gatzemeier, U.; Price, A.; Nicolson, M.; Mercier, R.; Mazabel, E.; Penning, C.; Zhang, M.H.; et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Dormán, G.; Cseh, S.; Hajdú, I.; Barna, L.; Kónya, D.; Kupai, K.; Kovács, L.; Ferdinandy, P. Matrix metalloproteinase inhibitors: A critical appraisal of design principles and proposed therapeutic utility. Drugs 2010, 70, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Amar, S.; Fields, G.B. Potential clinical implications of recent matrix metalloproteinase inhibitor design strategies. Expert Rev. Proteom. 2015, 12, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. New strategies for targeting matrix metalloproteinases. Matrix Biol. 2015, 44–46, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Rims, C.R.; McGuire, J.K. Matrilysin (MMP-7) catalytic activity regulates β-catenin localization and signaling activation in lung epithelial cells. Exp. Lung Res. 2014, 40, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Valacca, C.; Tassone, E.; Mignatti, P. TIMP-2 Interaction with MT1-MMP Activates the AKT Pathway and Protects Tumor Cells from Apoptosis. PLoS ONE 2015, 10, e0136797. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Flexner, C. HIV-protease inhibitors. N. Engl. J. Med. 1998, 338, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Riddler, S.A.; Haubrich, R.; Di Rienzo, A.G.; Peeples, L.; Powderly, W.G.; Klingman, K.L.; Garren, K.W.; George, T.; Rooney, J.F.; Brizz, B.; et al. AIDS Clinical Trials Group Study A5142 Team. Class-sparing regimens for initial treatment of HIV-1 infection. N. Engl. J. Med. 2008, 358, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Global Report: UNAIDS Report on the Global AIDS Epidemic 2010; UNAIDS: Geneva, Switzerland, 2010. [Google Scholar]
- André, P.; Groettrup, M.; Klenerman, P.; de Giuli, R.; Booth, B.L., Jr; Cerundolo, V.; Bonneville, M.; Jotereau, F.; Zinkernagel, R.M.; Lotteau, V. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation and T cell responses. Proc. Natl. Acad. Sci. USA 1998, 95, 13120–13124. [Google Scholar] [CrossRef] [PubMed]
- Gaedicke, S.; Firat-Geier, E.; Constantiniu, O.; Lucchiari-Hartz, M.; Freudenberg, M.; Galanos, C.; Niedermann, G. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: Induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002, 62, 6901–6908. [Google Scholar] [PubMed]
- Pajonk, F.; Himmelsbach, J.; Riess, K.; Sommer, A.; McBride, W.H. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radio-sensitization in non-HIV-associated human cancer cells. Cancer Res. 2002, 62, 5230–5235. [Google Scholar] [PubMed]
- Yang, Y.; Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Takeuchi, T.; Adachi, Y.; Kobayashi, M.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br. J Cancer 2006, 95, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Hampson, L.; Kitchener, H.C.; Hampson, I.N. Specific HIV protease inhibitors inhibit the ability of HPV16 E6 to degrade p53 and selectively kill E6-dependent cervical carcinoma cells in vitro. Antivir. Ther. 2006, 11, 813–825. [Google Scholar] [PubMed]
- De Barros, S.; Zakaroff-Girard, A.; Lafontan, M.; Galitzky, J.; Bourlier, V. Inhibition of human preadipocyte proteasomal activity by HIV protease inhibitors or specific inhibitor lactacystin leads to a defect in adipogenesis, which involves matrix metalloproteinase-9. J. Pharmacol. Exp. Ther. 2007, 320, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Caron, M.; Vidal, H.; Jan, V.; Auclair, M.; Vigouroux, C.; Luboinski, J.; Laville, M.; Maachi, M.; Girard, P.M.; et al. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet 2002, 359, 1026–1031. [Google Scholar] [CrossRef]
- Riddle, T.M.; Kuhel, D.G.; Woollett, L.A.; Fichtenbaum, C.J.; Hui, D.Y. HIV protease inhibitor induces fatty acid and sterol biosynthesis in liver and adipose tissues due to the accumulation of activated sterol regulatory element-binding proteins in the nucleus. J. Biol. Chem. 2001, 276, 37514–37519. [Google Scholar] [CrossRef] [PubMed]
- Lenhard, J.M.; Croom, D.K.; Weiel, J.E.; Winegar, D.A. HIV protease inhibitors stimulate hepatic triglyceride synthesis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2625–2629. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y. Effects of HIV protease inhibitor therapy on lipid metabolism. Prog. Lipid Res. 2003, 42, 81–92. [Google Scholar] [CrossRef]
- Tran, H.; Robinson, S.; Mikhailenko, I.; Strickland, D.K. Modulation of the LDL receptor and LRP levels by HIV protease inhibitors. J. Lipid. Res. 2003, 44, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Hruz, P.W.; Mueckler, M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J. Biol. Chem. 2000, 275, 20251–20254. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Jung, N.; Hofmann, A.; Cornely, O.A.; Wyen, C.; Fätkenheuer, G.; Hartmann, P. Nucleoside-free boosted double PI regimen: Significant CD4+ T-cell recovery in patients with poor immunologic response despite virologic suppression. Curr. HIV Res. 2008, 6, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Cassone, A.; De Bernardis, F.; Torosantucci, A.; Tacconelli, E.; Tumbarello, M.; Cauda, R. In vitro and in vivo anticandidal activity of human immunodeficiency virus protease inhibitors. J. Infect. Dis. 1999, 180, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Krischer, J.; Rutschmann, O.; Hirschel, B.; Vollenweider-Roten, S.; Saurat, J.H.; Pechère, M. Regression of Kaposi’s sarcoma during therapy with HIV-1 protease inhibitors: A prospective pilot study. J. Am. Acad. Dermatol. 1998, 38, 594–598. [Google Scholar] [CrossRef]
- Lebbé, C.; Blum, L.; Pellet, C.; Blanchard, G.; Vérola, O.; Morel, P.; Danne, O.; Calvo, F. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi’s sarcoma. AIDS 1998, 12, F45–F49. [Google Scholar] [CrossRef] [PubMed]
- Niehues, T.; Horneff, G.; Megahed, M.; Schroten, H.; Wahn, V. Complete regression of AIDS-related Kaposi’s sarcoma in a child treated with highly active antiretroviral therapy. AIDS 1999, 13, 1148–1149. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Bourboulia, D.; Wilkinson, J.; Hayes, P.; Cope, A.; Marcelin, A.G.; Calvez, V.; Gotch, F.; Boshoff, C.; Gazzard, B. Prospective study of the effects of antiretroviral therapy on Kaposi sarcoma—Associated herpesvirus infection in patients with and without Kaposi sarcoma. J. Acquir. Immune Defic. Syndr. 2002, 31, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Hallit, R.R.; Afridi, M.; Sison, R.; Szabela, M.E.; Bajaj, N.; Alchaa, R.; Hallit, S.; Awkar, N.; Boghossian, J.; Slim, J. AIDS-related lymphoma: Resolution with antiretroviral therapy alone. J. Int. Assoc. Provid. AIDS Care 2014, 13, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Cobucci, R.N.; Lima, P.H.; de Souza, P.C.; Costa, V.V.; Cornetta Mda, C.; Fernandes, J.V.; Gonçalves, A.K. Assessing the impact of HAART on the incidence of defining and non-defining AIDS cancers among patients with HIV/AIDS: A systematic review. J. Infect. Public Health 2015, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Heard, I.; Schmitz, V.; Costagliola, D.; Orth, G.; Kazatchkine, M.D. Early regression of cervical lesions in HIV-seropositive women receiving highly active antiretroviral therapy. AIDS 1998, 12, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Firnhaber, C.; Westreich, D.; Schulze, D.; Williams, S.; Siminya, M.; Michelow, P.; Levin, S.; Faesen, M.; Smith, J.S. Highly active antiretroviral therapy and cervical dysplasia in HIV-positive women in South Africa. J. Int. AIDS Soc. 2012, 15, e17382. [Google Scholar] [CrossRef] [PubMed]
- Adler, D.H.; Kakinami, L.; Modisenyane, T.; Tshabangu, N.; Mohapi, L.; De Bruyn, G.; Martinson, N.A.; Omar, T. Increased regression and decreased incidence of human papillomavirus-related cervical lesions among HIV-infected women on HAART. AIDS 2012, 26, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Soncini, E.; Zoncada, A.; Condemi, V.; Antoni, A.D.; Bocchialini, E.; Soregotti, P. Reduction of the risk of cervical intraepithelial neoplasia in HIV-infected women treated with highly active antiretroviral therapy. Acta Biomed. 2007, 78, 36–40. [Google Scholar] [PubMed]
- Minkoff, H.; Ahdieh, L.; Massad, L.S.; Anastos, K.; Watts, D.H.; Melnick, S.; Muderspach, L.; Burk, R.; Palefsky, J. The effect of highly active antiretroviral therapy on cervical cytologic changes associated with oncogenic HPV among HIV-infected women. AIDS 2001, 15, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Monini, P.; Sgadari, C.; Toschi, E.; Barillari, G.; Ensoli, B. Antitumour effects of antiretroviral therapy. Nat. Rev. Cancer 2004, 4, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Bani-Sadr, F.; Fournier, S.; Molina, J.M. Relapse of Kaposi’s sarcoma in HIV-infected patients switching from a protease inhibitor to a non-nucleoside reverse transcriptase inhibitor-based highly active antiretroviral therapy regimen. AIDS 2003, 17, 1580–1581. [Google Scholar] [CrossRef] [PubMed]
- Bono, C.; Karlin, L.; Harel, S.; Mouly, E.; Labaume, S.; Galicier, L.; Apcher, S.; Sauvageon, H.; Fermand, J.P.; Bories, J.C.; et al. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica 2012, 97, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Brüning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Gingelmaiers, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol. Ther. 2009, 8, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.A.; Guo, S.; Valdes-Albini, F. Nelfinavir induces liposarcoma apoptosis and cell cycle arrest by up-regulating sterol regulatory element binding protein-1. Anticancer Drugs 2006, 17, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Tomita, M.; Katano, H.; Yamamoto, N.; Ahmed, S.; Yamamoto, M.; Sata, T.; Mori, N.; Yamamoto, N. An HIV protease inhibitor, ritonavir targets the nuclear factor-kappaB and inhibits the tumor growth and infiltration of EBV-positive lymphoblastoid B cells. Int. J. Cancer 2009, 124, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Lopiccolo, J.; Tsurutani, J. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy and apoptosis in vitro and in vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Saito, T.; Bandobashi, K.; Yang, Y.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol. Cancer Ther. 2004, 3, 473–479. [Google Scholar] [PubMed]
- Jiang, W.; Mikochik, P.J.; Ra, J.H.; Lei, H.; Flaherty, K.T.; Winkler, J.D.; Spitz, F.R. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res. 2007, 67, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Timeus, F.; Crescenzio, N.; Doria, A.; Foglia, L.; Pagliano, S.; Ricotti, E.; Fagioli, F.; Tovo, P.A.; Cordero di Montezemolo, L. In vitro anti-neuroblastoma activity of saquinavir and its association with imatinib. Oncol Rep. 2012, 27, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ikezoe, T.; Takeuchi, T.; Adachi, Y.; Ohtsuki, Y.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 2005, 96, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Bajpai, R.; Sharma, H.; Heitmeier, M.; Jain, A.D.; Matulis, S.M.; Nooka, A.K.; Mishra, R.K.; Hruz, P.W.; Schiltz, G.E.; et al. Development of GLUT4-selective antagonists for multiple myeloma therapy. Eur. J. Med. Chem. 2017, 139, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Justesen, U.S. Protease inhibitor plasma concentrations in HIV antiretroviral therapy. Dan. Med. Bull. 2008, 55, 165–185. [Google Scholar] [PubMed]
- Barillari, G.; Iovane, A.; Bacigalupo, I.; Palladino, C.; Bellino, S.; Leone, P.; Monini, P.; Ensoli, B. Ritonavir or saquinavir impairs the invasion of cervical intraepithelial neoplasia cells via a reduction of MMP expression and activity. AIDS 2012, 26, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Brüning, A.; Vogel, M.; Mylonas, I.; Friese, K.; Burges, A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr. Cancer Drug Targets 2011, 11, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Hampson, L.; Maranga, I.O.; Masinde, M.S.; Oliver, A.W.; Batman, G.; He, X.; Desai, M.; Okemwa, P.M.; Stringfellow, H.; Martin-Hirsch, P.; et al. A Single-Arm, Proof-Of-Concept Trial of Lopimune (Lopinavir/Ritonavir) as a Treatment for HPV-Related Pre-Invasive Cervical Disease. PLoS ONE 2016, 11, e0147917. [Google Scholar] [CrossRef] [PubMed]
- Bacigalupo, I.; Palladino, C.; Leone, P.; Toschi, E.; Sgadari, C.; Ensoli, B.; Barillari, G. Inhibition of MMP-9 expression by ritonavir or saquinavir is associated with inactivation of the AKT/Fra-1 pathway in cervical intraepithelial neoplasia cells. Oncol. Lett. 2017, 13, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Toschi, E.; Sgadari, C.; Malavasi, L.; Bacigalupo, I.; Chiozzini, C.; Carlei, D.; Compagnoni, D.; Bellino, S.; Bugarini, R.; Falchi, M.; et al. Human immunodeficiency virus protease inhibitors reduce the growth of human tumors via a proteasome-independent block of angiogenesis and matrix metalloproteinases. Int. J. Cancer 2011, 128, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Sgadari, C.; Monini, P.; Barillari, G.; Ensoli, B. Use of HIV protease inhibitors to block Kaposi’s sarcoma and tumor growth. Lancet Oncol. 2003, 4, 537–547. [Google Scholar] [CrossRef]
- Sgadari, C.; Barillari, G.; Toschi, E.; Carlei, D.; Bacigalupo, I.; Baccarini, S.; Palladino, C.; Leone, P.; Bugarini, R.; Malavasi, L.; et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat. Med. 2002, 8, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, G.M.; Mastroianni, C.M.; Latronico, T.; Mengoni, F.; Fasano, A.; Lichtner, M.; Vullo, V.; Riccio, P. Anti-HIV drugs decrease the expression of matrix metalloproteinases in astrocytes and microglia. Brain 2004, 127, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Latronico, T.; Liuzzi, G.M.; Riccio, P.; Lichtner, M.; Mengoni, F.; D’Agostino, C.; Vullo, V.; Mastroianni, C.M. Antiretroviral therapy inhibits matrix metalloproteinase-9 from blood mononuclear cells of HIV-infected patients. AIDS 2007, 21, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Monini, P.; Sgadari, C.; Grosso, M.G.; Bellino, S.; Di Biagio, A.; Toschi, E.; Bacigalupo, I.; Sabbatucci, M.; Cencioni, G.; Salvi, E.; et al. For the Concerted Action on Kaposi’s Sarcoma. Clinical course of classic Kaposi’s sarcoma in HIV-negative patients treated with the HIV protease inhibitor indinavir. AIDS 2009, 23, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.Y.; Tsai, P.C.; Tseng, C.H.; Chen, Y.L.; Chang, L.S.; Lin, S.R. Inhibition of EGF/EGFR activation with naphtho[1,2-b] furan-4,5-dione blocks migration and invasion of MDA-MB-231 cells. Toxicol. In Vitro 2013, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.P.; Mathur, R.S.; Rust, P.F.; Young, R.C. Human papilloma virus (HPV)-E6/E7 and epidermal growth factor receptor (EGF-R) protein levels in cervical cancer and cervical intraepithelial neoplasia (CIN). Am. J. Reprod. Immunol. 2001, 46, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, B.I.; Steine, S.J.; Sandvei, R.; Molven, A.; Laerum, O.D. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: Frequent PIK3CA amplification and AKT phosphorylation. Int. J. Cancer 2006, 118, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Du, C.X.; Wang, Y. Expression of P-Akt, NFkappa B and their correlation with human papillomavirus infection in cervical carcinoma. Eur. J. Gynaecol. Oncol. 2012, 33, 274–277. [Google Scholar] [PubMed]
- Batchu, R.B.; Gruzdyn, O.V.; Bryant, C.S.; Qazi, A.M.; Kumar, S.; Chamala, S.; Kung, S.T.; Sanka, R.S.; Puttagunta, U.S.; Weaver, D.W.; et al. Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the RB/E2F-1 and AKT pathways. Pharmaceuticals 2014, 7, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Lopiccolo, J.; Dennis, P.A. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy 2008, 4, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.; Müller-Ide, H.; Rückrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Plastaras, J.P.; Vapiwala, N.; Ahmed, M.S.; Gudonis, D.; Cerniglia, G.J.; Feldman, M.D.; Frank, I.; Gupta, A.K. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 2008, 7, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a novel Akt inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.W.; Pan, M.R.; Hou, M.F.; Hung, W.C. Cyclooxygenase-2 up-regulates CCR7 expression via AKT-mediated phosphorylation and activation of Sp1 in breast cancer cells. J. Cell. Physiol. 2013, 228, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Luo, M.Q.; Xia, M.; Wu, Q.; Long, S.M.; Hu, Y.; Gao, G.C.; Yao, X.L.; He, M.; Su, H.; et al. Marine compound catunaregin inhibits angiogenesis through the modulation of phosphorylation of akt and eNOS in vivo and in vitro. Mar. Drugs 2014, 12, 2790–2801. [Google Scholar] [CrossRef] [PubMed]
- Bottsford-Miller, J.N.; Coleman, R.L.; Sook, A.K. Resistance and escape from anti-angiogenesis therapy: Clinical implications and future strategies. J. Clin. Oncol. 2012, 30, 4026–4034. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: Implications for radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef] [PubMed]
- Barillari, G.; Iovane, A.; Bacigalupo, I.; Labbaye, C.; Chiozzini, C.; Sernicola, L.; Quaranta, M.T.; Falchi, M.; Sgadari, C.; Ensoli, B. The HIV protease inhibitor indinavir down-regulates the expression of the pro-angiogenic MT1-MMP by human endothelial cells. Angiogenesis 2014, 7, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W.; Engelse, M.A.; Quax, P.H. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler. Tromb. Vasc. Biol. 2006, 26, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Hakeem, A.; Bhatti, S.; Cilingiroglu, M. The spectrum of atherosclerotic coronary artery disease in HIV patients. Curr. Atheroscler. Rep. 2010, 12, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Jarvis, R.M.; Allwood, J.W.; Batman, G.; Moore, R.E.; Marsden-Edwards, E.; Hampson, L.; Hampson, I.N.; Goodacre, R. Raman chemical mapping reveals site of action of HIV protease inhibitors in HPV16 E6 expressing cervical carcinoma cells. Anal. Bioanal. Chem. 2010, 398, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Batman, G.; Oliver, A.W.; Zehbe, I.; Richard, C.; Hampson, L.; Hampson, I.N. Lopinavir up-regulates expression of the antiviral protein ribonuclease L in human papillomavirus-positive cervical carcinoma cells. Antivir. Ther. 2011, 16, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Pils, S.; Joura, E.A. From the monovalent to the ninevalent HPV vaccine. Clin. Microbiol. Infect. 2015, 21, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Apgar, B.S.; Kaufman, A.J.; Bettcher, C.; Parker-Featherstone, E. Gynecologic procedures: Colposcopy, treatment of cervical intraepithelial neoplasia and endometrial assessment. Am. Fam. Phys. 2013, 87, 836–843. [Google Scholar]
- Sigfrid, L.; Murphy, G.; Haldane, V.; Chuah, F.L.H.; Ong, S.E.; Cervero-Liceras, F.; Watt, N.; Alvaro, A.; Otero-Garcia, L.; Balabanova, D.; et al. Integrating cervical cancer with HIV healthcare services: A systematic review. PLoS ONE 2017, 12, e0181156. [Google Scholar] [CrossRef] [PubMed]
- Armarnik, S.; Sheiner, E.; Piura, B.; Meirovitz, M.; Zlotnik, A.; Levy, A. Obstetric outcome following cervical conization. Arch. Gynecol. Obstet. 2011, 283, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, T.; Takeuchi, H.; Jimi, E.; Beppu, M.; Shinohara, M.; Shirasuna, K. Involvement of proteasomes in migration and matrix metalloproteinase-9 production of oral squamous cell carcinoma. Int. J. Cancer 1998, 77, 578–585. [Google Scholar] [CrossRef]
- Li, J.Y.; Huang, W.Q.; Tu, R.H.; Zhong, G.Q.; Luo, B.B.; He, Y. Resveratrol rescues hyperglycemia-induced endothelial dysfunction via activation of Akt. Acta Pharmacol. Sin. 2017, 38, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Makinoshima, H.; Umemura, S.; Suzuki, A.; Nakanishi, H.; Maruyama, A.; Udagawa, H.; Mimaki, S.; Matsumoto, S.; Niho, S.; Ishii, G.; et al. Metabolic determinants of sensitivity to phosphatidylinositol 3-kinase pathway inhibitor in small-cell lung carcinoma. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.; Roubaud, G.; Cazeau, A.L.; Mahammedi, H.; Vedrine, L.; Joly, F.; Mourey, L.; Pfister, C.; Goberna, A.; Lortal, B.; et al. Safety and efficacy of temsirolimus second line treatment for patients with recurrent bladder cancer. BMC Cancer 2018, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.Y.; Han, L.; Li, X.; Yang, A.V.; Lu, J.; Guan, S.; Li, H.; Yu, Y.; Zhao, Y.; Yang, J.; et al. Novel proteasome inhibitor delanzomib sensitizes cervical cancer cells to doxorubicin-induced apoptosis via stabilizing tumor suppressor proteins in the p53 pathway. Oncotarget 2017, 8, 114123–114135. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Li, J.; Mao, X. Dissecting bortezomib: Development, application, adverse effects and future direction. Curr. Pharm. Des. 2013, 19, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Akers, W.J.; Xu, B.; Lee, H.; Sudlow, G.P.; Fields, G.B.; Achilefu, S.; Edwards, W.B. Detection of MMP-2 and MMP-9 activity in vivo with a triple-helical peptide optical probe. Bioconjugate Chem. 2012, 23, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, N.; van Waarde, A.; Bischoff, R.; Oltenfreiter, R.; van de Wiele, C.; Dierckx, R.A.; Elsinga, P.H. Probes for non-invasive matrix metalloproteinase-targeted imaging with PET and SPECT. Curr. Pharm. Des. 2013, 19, 4647–4672. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Temma, T.; Hara, I.; Makino, A.; Kondo, N.; Ozeki, E.; Ono, M.; Saji, H. In vivo imaging of membrane type-1 matrix metalloproteinase with a novel activatable near-infrared fluorescence probe. Cancer Sci. 2014, 105, 1056–1062. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barillari, G.; Monini, P.; Sgadari, C.; Ensoli, B. The Impact of Human Papilloma Viruses, Matrix Metallo-Proteinases and HIV Protease Inhibitors on the Onset and Progression of Uterine Cervix Epithelial Tumors: A Review of Preclinical and Clinical Studies. Int. J. Mol. Sci. 2018, 19, 1418. https://doi.org/10.3390/ijms19051418
Barillari G, Monini P, Sgadari C, Ensoli B. The Impact of Human Papilloma Viruses, Matrix Metallo-Proteinases and HIV Protease Inhibitors on the Onset and Progression of Uterine Cervix Epithelial Tumors: A Review of Preclinical and Clinical Studies. International Journal of Molecular Sciences. 2018; 19(5):1418. https://doi.org/10.3390/ijms19051418
Chicago/Turabian StyleBarillari, Giovanni, Paolo Monini, Cecilia Sgadari, and Barbara Ensoli. 2018. "The Impact of Human Papilloma Viruses, Matrix Metallo-Proteinases and HIV Protease Inhibitors on the Onset and Progression of Uterine Cervix Epithelial Tumors: A Review of Preclinical and Clinical Studies" International Journal of Molecular Sciences 19, no. 5: 1418. https://doi.org/10.3390/ijms19051418
APA StyleBarillari, G., Monini, P., Sgadari, C., & Ensoli, B. (2018). The Impact of Human Papilloma Viruses, Matrix Metallo-Proteinases and HIV Protease Inhibitors on the Onset and Progression of Uterine Cervix Epithelial Tumors: A Review of Preclinical and Clinical Studies. International Journal of Molecular Sciences, 19(5), 1418. https://doi.org/10.3390/ijms19051418