From Structure to Phenotype: Impact of Collagen Alterations on Human Health


Abstract
1. Introduction
2. The ECM: Molecular and Structural Diversity
2.1. Chemical Composition and Mechanical Properties
2.2. ECM-Bound Growth and Secreted Factors
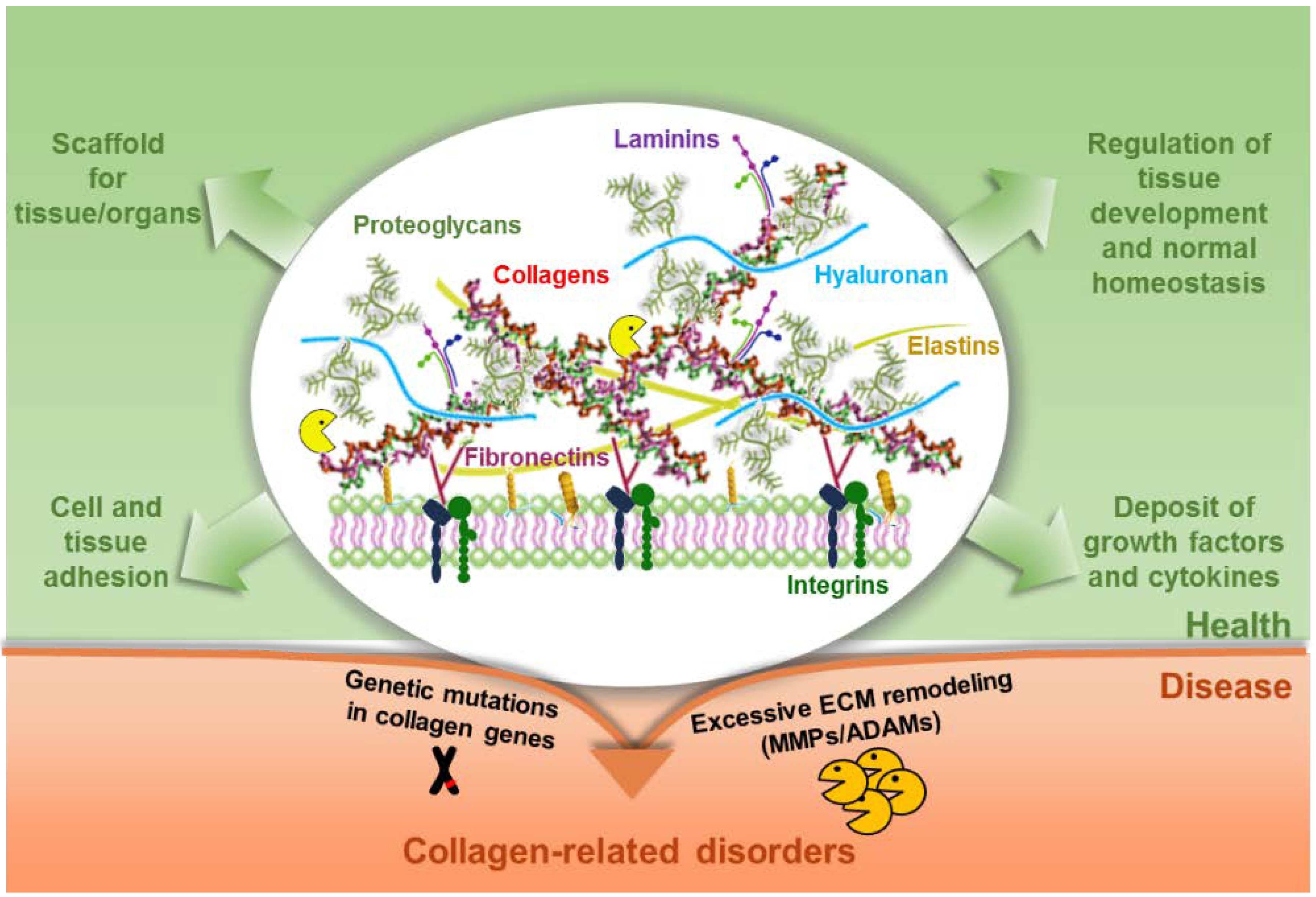
3. ECM Functions
3.1. Structural Roles of ECM
3.2. Signalling Modulation
3.3. ECM in Development
3.4. Cell Migration
3.5. ECM Remodelling
4. Collagens
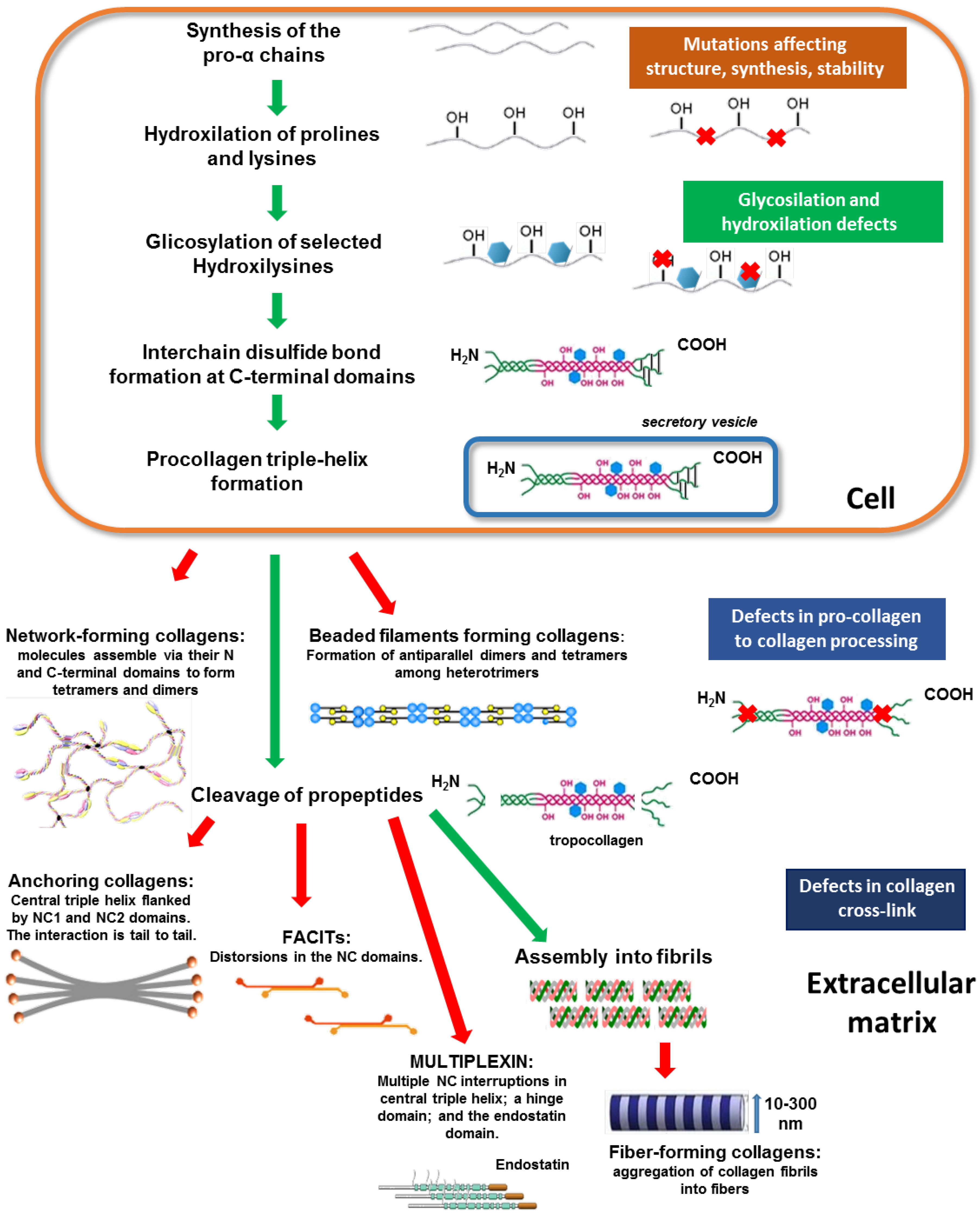
4.1. Collagen Synthesis and Organization
4.2. Nomenclature and Classification
4.3. Collagen Degradation
5. Collagen Alterations in Pathological Events
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Järveläinen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol Rev. 2009, 61, 198–223. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome-an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Almond, A. Hyaluronan. Cell. Mol. Life Sci. 2007, 64, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Abatangelo, G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999, 7, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Dicker, K.T.; Gurski, L.A.; Pradhan-Bhatt, S.; Witt, R.L.; Farach-Carson, M.C.; Jia, X. Hyaluronan: A simple polysaccharide with diverse biological functions. Acta Biomater. 2014, 10, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Volpi, N.; Schiller, J.; Stern, R.; Soltés, L. Role, metabolism, chemical modifications and applications of hyaluronan. Curr. Med. Chem. 2009, 16, 1718–1745. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, M. The laminin family. Cell. Adhes. Migr. 2013, 7, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.A. Extracellular Matrix; John Wiley & Sons Ltd.: Chichester, UK, 2001. [Google Scholar]
- Mithieux, S.M.; Weiss, A.S. Elastin. Adv. Protein Chem. 2005, 70, 437–461. [Google Scholar] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Wijelath, E.S.; Rahman, S.; Namekata, M.; Murray, J.; Nishimura, T.; Mostafavi-Pour, Z.; Patel, Y.; Suda, Y.; Humphries, M.J.; Sobel, M. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: Enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ. Res. 2006, 99, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Patel, Y.; Murray, J.; Patel, K.V.; Sumathipala, R.; Sobel, M.; Wijelath, E.S. Novel hepatocyte growth factor (HGF) binding domains on fibronectin and vitronectin coordinate a distinct and amplified Met-integrin induced signalling pathway in endothelial cells. Bmc Cell. Biol. 2005, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Ren, X.D.; Pan, Z.; Macri, L.; Zong, W.X.; Tonnesen, M.G.; Rafailovich, M.; Bar-Sagi, D.; Clark, R.A. Fibronectin growth factor-binding domains are required for fibroblast survival. J. Investig. Dermatol. 2011, 131, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Rifkin, D.B. Extracellular microfibrils: Contextual platforms for TGFβ and BMP signaling. Curr. Opin. Cell Biol. 2009, 21, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Yin, L.; Kothapalli, D.; Castagnino, P.; Byfield, F.J.; Xu, T.; Levental, I.; Hawthorne, E.; Janmey, P.A.; Assoian, R.K. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr. Biol. 2009, 19, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, C.; Vermeulen, P.; Van Marck, E.; Dirix, L. The presence of a fibrotic focus is an independent predictor of early metastasis in lymph node-negative breast cancer patients. Am. J. Surg. Pathol. 2001, 25, 1557–1558. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xie, T. Stem cell niche: Structure and function. Annu. Rev. Cell Dev. Biol. 2005, 21, 605–631. [Google Scholar] [CrossRef] [PubMed]
- Taddei, I.; Deugnier, M.A.; Faraldo, M.M.; Petit, V.; Bouvard, D.; Medina, D.; Fässler, R.; Thiery, J.P.; Glukhova, M.A. Beta1 integrin deletion from the basal compartment of the mammary epithelium affects stem cells. Nat. Cell Biol. 2008, 10, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Lin, X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb. Perspect. Biol. 2009, 1, a002493. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Salza, R. Matricryptins and matrikines: Biologically active fragments of the extracellular matrix. Exp. Dermatol. 2014, 23, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Maquart, F.X.; Siméon, A.; Pasco, S.; Monboisse, J.C. Regulation of cell activity by the extracellular matrix: The concept of matrikines. J. Soc. Biol. 1999, 193, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Ballut, L. Matricryptins derived from collagens and proteoglycans. Front. Biosci. 2011, 16, 674–697. [Google Scholar] [CrossRef]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Väisänen, M.R.; Väisänen, T.; Pihlajaniemi, T. The shed ectodomain of type XIII collagen affects cell behaviour in a matrix-dependent manner. Biochem. J. 2004, 380, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.M.; Simons, M. The extracellular matrix and blood vessel formation: Not just a scaffold. J. Cell. Mol. Med. 2007, 11, 176–205. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Stenbjorn, R.S.; Gorse, K.; Su, K.; Hauser, K.F.; Ricard-Blum, S.; Pihlajaniemi, T.; Fox, M.A. Target-derived matricryptins organize cerebellar synapse formation through α3β1 integrins. Cell Rep. 2012, 2, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.L.; Valiathan, R.R.; Arkwright, R.; Sohail, A.; Mihai, C.; Kumarasiri, M.; Mahasenan, K.V.; Mobashery, S.; Huang, P.; Agarwal, G.; et al. Discoidin domain receptors: Unique receptor tyrosine kinases in collagen-mediated signaling. J. Biol. Chem. 2013, 288, 7430–7437. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.H. Extracellular matrix in development: Insights from mechanisms conserved between invertebrates and vertebrates. Cold Spring Harb. Perspect. Biol. 2011, 3, a005082. [Google Scholar] [CrossRef] [PubMed]
- Aiken, A.; Khokha, R. Unraveling metalloproteinase function in skeletal biology and disease using genetically altered mice. Biochim. Biophys. Acta Mol. Cell. Res. 2010, 1803, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Ballestrem, C.; Kam, Z.; Geiger, B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 2003, 116, 4605–4613. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Danen, E.H. Adhesion signaling- crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Fasciglione, G.F.; Gioia, M.; Ciaccio, C.; Tundo, G.R.; Marini, S.; Coletta, M. Human matrix metalloproteinases: An ubiquitarian class of enzymes involved in several pathological processes. Mol. Asp. Med. 2012, 33, 119–208. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. The adams: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.S. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: Functions and mechanisms. J. Biol. Chem. 2009, 284, 31493–31497. [Google Scholar] [CrossRef] [PubMed]
- Bertenshaw, G.P.; Norcum, M.T.; Bond, J.S. Structure of homo- and hetero-oligomeric meprin metalloproteases. Dimers, tetramers, and high molecular mass multimers. J. Biol. Chem. 2003, 278, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, A.; Legrand, C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb. Res. 2000, 98, 323–332. [Google Scholar] [CrossRef]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol. 2006, 38, 2018–2039. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, K.; Morimoto-Tomita, M.; Bistrup, A.; Li, J.; Lyon, M.; Gallagher, J.; Werb, Z.; Rosen, S.D. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: Effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Bastow, E.R.; Byers, S.; Golub, S.B.; Clarkin, C.E.; Pitsillides, A.A.; Fosang, A.J. Hyaluronan synthesis and degradation in cartilage and bone. Cell. Mol. Life Sci. 2008, 65, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Raines, R.T. Modulating collagen triple-helix stability with 4-chloro, 4-fluoro, and 4-methylprolines. Adv. Exp. Med. Biol. 2009, 611, 251–252. [Google Scholar] [PubMed]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A. Biochemistry of Collagens, Laminins and Elastin: Structure, Function and Biomarkers; Elsevier: New York, NY, USA, 2016. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Collagen: The Fibrous Proteins of the Matrix. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000; Section 22.3. [Google Scholar]
- Canty, E.G.; Kadler, K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005, 118, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Carrique, L.; Vadon-Le Goff, S.; Mariano, N.; Georges, R.N.; Delolme, F.; Koivunen, P.; Myllyharju, J.; Moali, C.; Aghajari, N.; et al. Structural basis of homo- and heterotrimerization of collagen I. Nat. Commun. 2017, 8, 14671. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, G.; Li, Y.; Mohs, A.; Strafaci, C.; Popiel, M.; Baum, J.; Brodsky, B. Common interruptions in the repeating tripeptide sequence of non-fibrillar collagens: Sequence analysis and structural studies on triple-helix peptide models. J. Mol. Biol. 2008, 376, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.R.; Paz, M.A.; Gallop, P.M. Cross-linking in collagen and elastin. Annu. Rev. Biochem. 1984, 53, 717–748. [Google Scholar] [CrossRef] [PubMed]
- Knupp, C.; Squire, J.M. Molecular packing in network-forming collagens. Adv. Protein Chem. 2005, 70, 375–403. [Google Scholar] [PubMed]
- Franzke, C.W.; Tasanen, K.; Schumann, H.; Bruckner-Tuderman, L. Collagenous transmembrane proteins: Collagen XVII as a prototype. Matrix Biol. 2003, 22, 299–309. [Google Scholar] [CrossRef]
- Fratzl, P. Collagen: Structure and Mechanics; Springer: New York, NY, USA, 2008. [Google Scholar]
- Marneros, A.G.; Olsen, B.R. Physiological role of collagen XVIII and endostatin. FASEB J. 2005, 19, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Marinkovich, M.P.; Veis, A.; Cai, X.; Rao, C.N.; O’Toole, E.A.; Woodley, D.T. Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J. Biol. Chem. 1997, 272, 14516–14522. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, J.K.; van Golde, L.M.; Batenburg, J.J. Collectins: Players of the innate immune system. Eur. J. Biochem. 2004, 271, 1229–1249. [Google Scholar] [CrossRef] [PubMed]
- Everts, V.; van der Zee, E.; Creemers, L.; Beertsen, W. Phagocytosis and intracellular digestion of collagen, its role in turnover and remodelling. Histochem. J. 1996, 28, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Parks, W.C.; Mecham, R.P. Extracellular Matrix Degradation; Springer: Berlin, Germany; London, UK, 2011. [Google Scholar]
- Monea, S.; Lehti, K.; Keski-Oja, J.; Mignatti, P. Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J. Cell. Physiol. 2002, 192, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.D.; Wang, Y.; Bresnick, A.; Dawson, J.; Janmey, P.A.; McCulloch, C.A. Collagen remodeling by phagocytosis is determined by collagen substrate topology and calcium-dependent interactions of gelsolin with nonmuscle myosin IIA in cell adhesions. Mol. Biol. Cell 2013, 24, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Shingleton, W.D.; Hodges, D.J.; Brick, P.; Cawston, T.E. Collagenase: A key enzyme in collagen turnover. Biochem. Cell Biol. 1996, 74, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Ingvarsen, S.; Jürgensen, H.J.; Melander, M.C.; Kjøller, L.; Moyer, A.; Honoré, C.; Madsen, C.A.; Garred, P.; Burgdorf, S.; et al. The non-phagocytic route of collagen uptake: A distinct degradation pathway. J. Biol. Chem. 2011, 286, 26996–27010. [Google Scholar] [CrossRef] [PubMed]
- Muschler, J.; Streuli, C.H. Cell-matrix interactions in mammary gland development and breast cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003202. [Google Scholar] [CrossRef] [PubMed]
- Barat-Houari, M.; Sarrabay, G.; Gatinois, V.; Fabre, A.; Dumont, B.; Genevieve, D.; Touitou, I. Mutation update for COL2A1 gene variants associated with type II collagenopathies. Hum. Mutat. 2016, 37, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Wenstrup, R.; De Paepe, A. Ehlers-Danlos Syndrome, Classic Type. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2007. [Google Scholar]
- Yeowell, H.N.; Steinmann, B. Ehlers-Danlos Syndrome, Kyphoscoliotic Form. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2000. [Google Scholar]
- Müller, T.; Mizumoto, S.; Suresh, I.; Komatsu, Y.; Vodopiutz, J.; Dundar, M.; Straub, V.; Lingenhel, A.; Melmer, A.; Lechner, S.; et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 2013, 22, 3761–3772. [Google Scholar] [CrossRef] [PubMed]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170A, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Dordoni, C.; Cinquina, V.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic Ehlers-Danlos syndrome. Orphanet J. Rare Dis. 2017, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; De Coster, P.; Hausser, I.; van Essen, A.J.; Franck, P.; Colige, A.; Nusgens, B.; Martens, L.; De Paepe, A. The natural history, including orofacial features of three patients with Ehlers-Danlos syndrome, dermatosparaxis type (EDS type VIIC). Am. J. Med. Genet. A 2004, 131, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, F.S.; Sillence, D.O. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A 2014, 164A, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Forlino, A.; Bächinger, H.P.; Bishop, N.J.; Byers, P.H.; Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 2017, 3, 17052. [Google Scholar] [CrossRef] [PubMed]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006, 127, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Fratzl-Zelman, N.; Barnes, A.M.; Weis, M.; Carter, E.; Hefferan, T.E.; Perino, G.; Chang, W.; Smith, P.A.; Roschger, P.; Klaushofer, K.; et al. Non-lethal type VIII osteogenesis imperfecta has elevated bone matrix mineralization. J. Clin. Endocrinol. Metab. 2016, 101, 3516–3525. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, F.S.; Nesbitt, I.M.; Zwikstra, E.H.; Nikkels, P.G.; Piersma, S.R.; Fratantoni, S.A.; Jimenez, C.R.; Huizer, M.; Morsman, A.C.; Cobben, J.M.; et al. Ppib mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.J.; Byers, P.H.; Glorieux, F.H.; Sponsellor, P.D. Osteogenesis Imperfecta: A Translational Approach to Brittle Bone Disease; Academic Press: Cambridge, MA, USA, 2014; ISBN 978-0-12-397165-4. [Google Scholar]
- Kelley, B.P.; Malfait, F.; Bonafe, L.; Baldridge, D.; Homan, E.; Symoens, S.; Willaert, A.; Elcioglu, N.; Van Maldergem, L.; Verellen-Dumoulin, C.; et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and bruck syndrome. J. Bone Miner. Res. 2011, 26, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Londono, R.; Fahiminiya, S.; Majewski, J.; Tétreault, M.; Nadaf, J.; Kannu, P.; Sochett, E.; Howard, A.; Stimec, J.; Dupuis, L.; et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet. 2015, 96, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Kamoun-Goldrat, A.; le Merrer, M. Infantile cortical hyperostosis (Caffey disease): A review. J. Oral Maxillofac. Surg. 2008, 66, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Meigel, W.N.; Müller, P.K.; Pontz, B.F.; Sörensen, N.; Spranger, J. A constitutional disorder of connective tissue suggesting a defect in collagen biosynthesis. Klin. Wochenschr. 1974, 52, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Anderson, I.J.; Goldberg, R.B.; Marion, R.W.; Upholt, W.B.; Tsipouras, P. Spondyloepiphyseal dysplasia congenita: Genetic linkage to type II collagen (COL2AI). Am. J. Hum. Genet. 1990, 46, 896–901. [Google Scholar] [PubMed]
- Nishimura, G.; Saitoh, Y.; Okuzumi, S.; Imaizumi, K.; Hayasaka, K.; Hashimoto, M. Spondyloepiphyseal dysplasia with accumulation of glycoprotein in the chondrocytes: Spondyloepiphyseal dysplasia, stanescu type. Skelet. Radiol. 1998, 27, 188–194. [Google Scholar] [CrossRef]
- Hammarsjö, A.; Nordgren, A.; Lagerstedt-Robinson, K.; Malmgren, H.; Nilsson, D.; Wedrén, S.; Nordenskjöld, M.; Nishimura, G.; Grigelioniene, G. Pathogenenic variant in the COL2A1 gene is associated with Spondyloepiphyseal dysplasia type Stanescu. Am. J. Med. Genet. A 2016, 170A, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; Goldberg, L.; Hof, J.O. Dominant inheritance of multiple epiphyseal dysplasia, myopia and deafness. Clin. Genet. 1978, 14, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Comstock, J.M.; Putnam, A.R.; Sangle, N.; Lowichik, A.; Rose, N.C.; Opitz, J.M. Recurrence of achondrogenesis type 2 in sibs: Additional evidence for germline mosaicism. Am. J. Med. Genet. A 2010, 152A, 1822–1824. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, K.; Marik, I.; Marikova, O.; Zemkova, D.; Kuklik, M. Czech dysplasia metatarsal type. Am. J. Med. Genet. A 2004, 129A, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hoornaert, K.P.; Marik, I.; Kozlowski, K.; Cole, T.; Le Merrer, M.; Leroy, J.G.; Coucke, P.J.; Sillence, D.; Mortier, G.R. Czech dysplasia metatarsal type: Another type II collagen disorder. Eur. J. Hum. Genet. 2007, 15, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.M.; Liu, Y.F.; Lin, M.W.; Chen, I.C.; Lin, P.Y.; Lin, G.L.; Jou, Y.S.; Lin, Y.T.; Fann, C.S.; Wu, J.Y.; et al. Autosomal dominant avascular necrosis of femoral head in two Taiwanese pedigrees and linkage to chromosome 12q13. Am. J. Hum. Genet. 2004, 75, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, R.G.; Katzenstein, P.L.; Moskowitz, R.W.; Weaver, E.J.; Malemud, C.J.; Pathria, M.N.; Jimenez, S.A.; Prockop, D.J. Genetic linkage of a polymorphism in the type II procollagen gene (COL2A1) to primary osteoarthritis associated with mild chondrodysplasia. N. Engl. J. Med. 1990, 322, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Learmonth, I.D.; Christy, G.; Beighton, P. Namaqualand hip dysplasia. Orthopedic implications. Clin. Orthop. Relat. Res. 1987, 218, 142–147. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein vapb causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Zankl, A.; Neumann, L.; Ignatius, J.; Nikkels, P.; Schrander-Stumpel, C.; Mortier, G.; Omran, H.; Wright, M.; Hilbert, K.; Bonafé, L.; et al. Dominant negative mutations in the C-propeptide of COL2A1 cause platyspondylic lethal skeletal dysplasia, torrance type, and define a novel subfamily within the type 2 collagenopathies. Am. J. Med. Genet. A 2005, 133A, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Neumann, L.; Kunze, J.; Uhl, M.; Stöver, B.; Zabel, B.; Spranger, J. Survival to adulthood and dominant inheritance of platyspondylic skeletal dysplasia, Torrance-Luton type. Pediatr. Radiol. 2003, 33, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Zankl, A.; Scheffer, H.; Schinzel, A. Ectodermal dysplasia with tetramelic deficiencies and no mutation in p63: Odontotrichomelic syndrome or a new entity? Am. J. Med. Genet. A 2004, 127A, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.E.; Sillence, D.O.; Lachman, R.S.; Toomey, K.; Bull, M.; Dorst, J.; Rimoin, D.L. Spondylometepiphyseal dysplasia, strudwick type. Am. J. Med. Genet. 1982, 13, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Tiller, G.E.; Polumbo, P.A.; Weis, M.A.; Bogaert, R.; Lachman, R.S.; Cohn, D.H.; Rimoin, D.L.; Eyre, D.R. Dominant mutations in the type II collagen gene, COL2A1, produce spondyloepimetaphyseal dysplasia, strudwick type. Nat. Genet. 1995, 11, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.; Booth, C.; Fillman, C.; Shapiro, M.; Blair, M.P.; Hyland, J.C.; Ala-Kokko, L. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am. J. Med. Genet. A 2011, 155A, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Faber, J.; Winterpacht, A.; Zabel, B.; Gnoinski, W.; Schinzel, A.; Steinmann, B.; Superti-Furga, A. Clinical variability of stickler syndrome with a COL2A1 haploinsufficiency mutation: Implications for genetic counselling. J. Med. Genet. 2000, 37, 318–320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Majava, M.; Hoornaert, K.P.; Bartholdi, D.; Bouma, M.C.; Bouman, K.; Carrera, M.; Devriendt, K.; Hurst, J.; Kitsos, G.; Niedrist, D.; et al. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am. J. Med. Genet. A 2007, 143A, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Vikkula, M.; Mariman, E.C.; Lui, V.C.; Zhidkova, N.I.; Tiller, G.E.; Goldring, M.B.; van Beersum, S.E.; de Waal Malefijt, M.C.; van den Hoogen, F.H.; Ropers, H.H. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 1995, 80, 431–437. [Google Scholar] [CrossRef]
- Nikopoulos, K.; Schrauwen, I.; Simon, M.; Collin, R.W.; Veckeneer, M.; Keymolen, K.; Van Camp, G.; Cremers, F.P.; van den Born, L.I. Autosomal recessive Stickler syndrome in two families is caused by mutations in the COL9A1 gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4774–4779. [Google Scholar] [CrossRef] [PubMed]
- McAlinden, A.; Majava, M.; Bishop, P.N.; Perveen, R.; Black, G.C.; Pierpont, M.E.; Ala-Kokko, L.; Männikkö, M. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of stickler syndrome. Hum. Mutat. 2008, 29, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Malizos, K.N.; Karantanas, A.H.; Varitimidis, S.E.; Dailiana, Z.H.; Bargiotas, K.; Maris, T. Osteonecrosis of the femoral head: Etiology, imaging and treatment. Eur. J. Radiol. 2007, 63, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Gilbert-Barnes, E.; Langer, L.O.; Opitz, J.M.; Laxova, R.; Sotelo-Arila, C. Kniest dysplasia: Radiologic, histopathological, and scanning electronmicroscopic findings. Am. J. Med. Genet. 1996, 63, 34–45. [Google Scholar] [CrossRef]
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Crovetto, F.; Moroni, G.; Zaina, B.; Acaia, B.; Ossola, M.W.; Fedele, L. Pregnancy in women with Alport syndrome. Int. Urol. Nephrol. 2013, 45, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Anker, M.C.; Arnemann, J.; Neumann, K.; Ahrens, P.; Schmidt, H.; König, R. Alport syndrome with diffuse leiomyomatosis. Am. J. Med. Genet. A 2003, 119A, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Longo, I.; Porcedda, P.; Mari, F.; Giachino, D.; Meloni, I.; Deplano, C.; Brusco, A.; Bosio, M.; Massella, L.; Lavoratti, G.; et al. COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 2002, 61, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Zenteno, J.C.; Crespí, J.; Buentello-Volante, B.; Buil, J.A.; Bassaganyas, F.; Vela-Segarra, J.I.; Diaz-Cascajosa, J.; Marieges, M.T. Next generation sequencing uncovers a missense mutation in col4a1 as the cause of familial retinal arteriolar tortuosity. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, E.; Chen, Z.; Gekeler, F.; Benhassine, S.; Dahan, K.; Marro, B.; Alamowitch, S.; Paques, M.; Ronco, P. Novel COL4A1 mutations associated with HANAC syndrome: A role for the triple helical CB3[IV] domain. Am. J. Med. Genet. A 2010, 152A, 2550–2555. [Google Scholar] [CrossRef] [PubMed]
- Lanfranconi, S.; Markus, H.S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: A systematic review. Stroke 2010, 41, e513–e518. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, Y.; Haginoya, K.; Kato, M.; Osaka, H.; Yokochi, K.; Arai, H.; Kakita, A.; Yamamoto, T.; Otsuki, Y.; Shimizu, S.; et al. Phenotypic spectrum of COL4A1 mutations: Porencephaly to schizencephaly. Ann. Neurol. 2013, 73, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Granata, T.; Freri, E.; Caccia, C.; Setola, V.; Taroni, F.; Battaglia, G. Schizencephaly: Clinical spectrum, epilepsy, and pathogenesis. J. Child Neurol. 2005, 20, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.M.; Singh, I.P.; Gandhi, C.D.; Prestigiacomo, C.J. Genetic risk factors for spontaneous intracerebral haemorrhage. Nat. Rev. Neurol. 2016, 12, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Bach, E.; Neuner, C.; Nanda, I.; Dysek, S.; Bittner, R.E.; Keller, A.; Bartsch, O.; Mlynski, R.; Haaf, T.; et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur. J. Hum. Genet. 2014, 22, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Badenas, C.; Praga, M.; Tazón, B.; Heidet, L.; Arrondel, C.; Armengol, A.; Andrés, A.; Morales, E.; Camacho, J.A.; Lens, X.; et al. Mutations in the COL4A4 and COL4A3 genes cause familial benign hematuria. J. Am. Soc. Nephrol. 2002, 13, 1248–1254. [Google Scholar] [PubMed]
- Bertini, E.; Pepe, G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur. J. Paediatr. Neurol. 2002, 6, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Nishino, I. Ullrich congenital muscular dystrophy: Clinicopathological features, natural history and pathomechanism(s). J. Neurol. Neurosurg. Psychiatry 2015, 86, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Martoni, E.; Grumati, P.; Sabatelli, P.; Squarzoni, S.; Urciuolo, A.; Ferlini, A.; Gualandi, F.; Bonaldo, P. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 2008, 71, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Zech, M.; Lam, D.D.; Francescatto, L.; Schormair, B.; Salminen, A.V.; Jochim, A.; Wieland, T.; Lichtner, P.; Peters, A.; Gieger, C.; et al. Recessive mutations in the α3 (VI) collagen gene COL6A3 cause early-onset isolated dystonia. Am. J. Hum. Genet. 2015, 96, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Rashidghamat, E.; McGrath, J.A. Novel and emerging therapies in the treatment of recessive dystrophic epidermolysis bullosa. Intractable Rare Dis Res. 2017, 6, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Titeux, M.; Pendaries, V.; Tonasso, L.; Décha, A.; Bodemer, C.; Hovnanian, A. A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum. Mutat. 2008, 29, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sato-Matsumura, K.C.; Yasukawa, K.; Tomita, Y.; Shimizu, H. Toenail dystrophy with COL7A1 glycine substitution mutations segregates as an autosomal dominant trait in 2 families with dystrophic epidermolysis bullosa. Arch. Dermatol. 2002, 138, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Baratz, K.H.; Tosakulwong, N.; Ryu, E.; Brown, W.L.; Branham, K.; Chen, W.; Tran, K.D.; Schmid-Kubista, K.E.; Heckenlively, J.R.; Swaroop, A.; et al. E2-2 protein and Fuchs’s corneal dystrophy. N. Engl. J. Med. 2010, 363, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Gottsch, J.D.; Sundin, O.H.; Liu, S.H.; Jun, A.S.; Broman, K.W.; Stark, W.J.; Vito, E.C.; Narang, A.K.; Thompson, J.M.; Magovern, M. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of fuchs corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Munier, F.L.; Yardley, J.; Hart-Holden, N.; Perveen, R.; Cousin, P.; Sutphin, J.E.; Noble, B.; Batterbury, M.; Kielty, C.; et al. Missense mutations in COL8A2, the gene encoding the α2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 2001, 10, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.C.; Marcus-Soekarman, D.; Stolte-Dijkstra, I.; Verrips, A.; Taylor, J.A.; Briggs, M.D. Type IX collagen gene mutations can result in multiple epiphyseal dysplasia that is associated with osteochondritis dissecans and a mild myopathy. Am. J. Med. Genet. A 2010, 152A, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Muragaki, Y.; Mundlos, S.; Upton, J.; Olsen, B.R. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science 1996, 272, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Czarny-Ratajczak, M.; Lohiniva, J.; Rogala, P.; Kozlowski, K.; Perälä, M.; Carter, L.; Spector, T.D.; Kolodziej, L.; Seppänen, U.; Glazar, R.; et al. A mutation in COL9A1 causes multiple epiphyseal dysplasia: Further evidence for locus heterogeneity. Am. J. Hum. Genet. 2001, 69, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, O.; Susic, M.; Ward, L.; Barclay, C.; Glorieux, F.H.; Cole, W.G. Schmid type of metaphyseal chondrodysplasia and COL10A1 mutations—findings in 10 patients. Am. J. Med. Genet. A 2005, 137A, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Çalışkan, E.; Açıkgöz, G.; Yeniay, Y.; Özmen, İ.; Gamsızkan, M.; Akar, A. A case of Marshall’s syndrome and review of the literature. Int. J. Dermatol. 2015, 54, e217–e221. [Google Scholar] [CrossRef] [PubMed]
- Tompson, S.W.; Faqeih, E.A.; Ala-Kokko, L.; Hecht, J.T.; Miki, R.; Funari, T.; Funari, V.A.; Nevarez, L.; Krakow, D.; Cohn, D.H. Dominant and recessive forms of fibrochondrogenesis resulting from mutations at a second locus, COL11A2. Am. J. Med. Genet. A 2012, 158A, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Tomek, M.S.; Van Laer, L.; Smith, S.; Kenyon, J.B.; Van Camp, G.; Smith, R.J. A novel locus for autosomal dominant nonsyndromic hearing loss, DFNA13, maps to chromosome 6p. Am. J. Hum. Genet. 1997, 61, 924–927. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Steensel, M.A.; Buma, P.; de Waal Malefijt, M.C.; van den Hoogen, F.H.; Brunner, H.G. Oto- spondylo-megaepiphyseal dysplasia (OSMED): Clinical description of three patients homozygous for a missense mutation in the COL11A2 gene. Am. J. Med. Genet. 1997, 70, 315–323. [Google Scholar] [CrossRef]
- Logan, C.V.; Cossins, J.; Rodríguez Cruz, P.M.; Parry, D.A.; Maxwell, S.; Martínez-Martínez, P.; Riepsaame, J.; Abdelhamed, Z.A.; Lake, A.V.; Moran, M.; et al. Congenital myasthenic syndrome type 19 is caused by mutations in COL13A1, encoding the atypical non-fibrillar collagen type XIII α1 chain. Am. J. Hum. Genet. 2015, 97, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Oliver, V.F.; van Bysterveldt, K.A.; Cadzow, M.; Steger, B.; Romano, V.; Markie, D.; Hewitt, A.W.; Mackey, D.A.; Willoughby, C.E.; Sherwin, T.; et al. A COL17A1 splice-altering mutation is prevalent in inherited recurrent corneal erosions. Ophthalmology 2016, 123, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Khan, A.O.; Mohamed, J.; Alkuraya, F.S. Novel recessive BFSP2 and PITX3 mutations: Insights into mutational mechanisms from consanguineous populations. Genet. Med. 2011, 13, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Shinwari, J.M.; Khan, A.; Awad, S.; Shinwari, Z.; Alaiya, A.; Alanazi, M.; Tahir, A.; Poizat, C.; Al Tassan, N. Recessive mutations in COL25A1 are a cause of congenital cranial dysinnervation disorder. Am. J. Hum. Genet. 2015, 96, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Ramirez, N.; Betz, R.; Mulcahey, M.J.; Pino, F.; Herrera-Soto, J.A.; Carlo, S.; Cornier, A.S. Steel syndrome: Dislocated hips and radial heads, carpal coalition, scoliosis, short stature, and characteristic facial features. J. Pediatr. Orthop. 2010, 30, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Intong, L.R.; Murrell, D.F. Inherited epidermolysis bullosa: New diagnostic criteria and classification. Clin. Dermatol. 2012, 30, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Saratzis, A.; Sarafidis, P.; Melas, N.; Khaira, H. Comparison of the impact of open and endovascular abdominal aortic aneurysm repair on renal function. J. Vasc. Surg. 2014, 60, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Orioli, D.; Compe, E.; Nardo, T.; Mura, M.; Giraudon, C.; Botta, E.; Arrigoni, L.; Peverali, F.A.; Egly, J.M.; Stefanini, M. XPD mutations in trichothiodystrophy hamper collagen VI expression and reveal a role of TFIIH in transcription derepression. Hum. Mol. Genet. 2013, 22, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Arseni, L.; Lanzafame, M.; Compe, E.; Fortugno, P.; Afonso-Barroso, A.; Peverali, F.A.; Lehmann, A.R.; Zambruno, G.; Egly, J.M.; Stefanini, M.; et al. TFIIH-dependent MMP-1 overexpression in trichothiodystrophy leads to extracellular matrix alterations in patient skin. Proc. Natl. Acad. Sci. USA 2015, 112, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, M.; Botta, E.; Lanzafame, M.; Orioli, D. Trichothiodystrophy: From basic mechanisms to clinical implications. DNA Repair 2010, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Faghri, S.; Tamura, D.; Kraemer, K.H.; Digiovanna, J.J. Trichothiodystrophy: A systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J. Med. Genet. 2008, 45, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Söderhäll, C.; Marenholz, I.; Kerscher, T.; Rüschendorf, F.; Esparza-Gordillo, J.; Worm, M.; Gruber, C.; Mayr, G.; Albrecht, M.; Rohde, K.; et al. Variants in a novel epidermal collagen gene (COL29A1) are associated with atopic dermatitis. PLoS Biol. 2007, 5, e242. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Gawkrodger, D.J.; Mortimer, M.J.; Jaron, A.G. The prevalence of childhood atopic eczema in a general population. J. Am. Acad. Dermatol. 1994, 30, 35–39. [Google Scholar] [CrossRef]
- Moravej, H.; Karamifar, H.; Karamizadeh, Z.; Amirhakimi, G.; Atashi, S.; Nasirabadi, S. Bruck syndrome—A rare syndrome of bone fragility and joint contracture and novel homozygous FKBP10 mutation. Endokrynol. Polska 2015, 66, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Ha-Vinh, R.; Alanay, Y.; Bank, R.A.; Campos-Xavier, A.B.; Zankl, A.; Superti-Furga, A.; Bonafé, L. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am. J. Med. Genet. A 2004, 131, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Giunta, C.; Krabichler, B.; Rüschendorf, F.; Zoppi, N.; Colombi, M.; Bittner, R.E.; Quijano-Roy, S.; Muntoni, F.; Cirak, S.; et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am. J. Hum. Genet. 2012, 90, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Avgitidou, G.; Siebelmann, S.; Bachmann, B.; Kohlhase, J.; Heindl, L.M.; Cursiefen, C. Brittle cornea syndrome: Case report with novel mutation in the PRDM5 gene and review of the literature. Case Rep. Ophthalmol. Med. 2015, 2015, 637084. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morava, E.; Guillard, M.; Lefeber, D.J.; Wevers, R.A. Autosomal recessive cutis laxa syndrome revisited. Eur. J. Hum. Genet. 2009, 17, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Yeung, C.Y.; Liu, H.L.; Ho, C.S.; Shu, C.H.; Chuang, C.K.; Huang, Y.W.; Wu, T.Y.; Huang, Z.D.; Jian, Y.R.; et al. A novel mutation in PYCR1 causes an autosomal recessive cutis laxa with premature aging features in a family. Am. J. Med. Genet. A 2011, 155A, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Kivuva, E.C.; Parker, M.J.; Cohen, M.C.; Wagner, B.E.; Sobey, G. De barsy syndrome: A review of the phenotype. Clin. Dysmorphol. 2008, 17, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Zirnsak, B.; Escande-Beillard, N.; Ganesh, J.; Tan, Y.X.; Al Bughaili, M.; Lin, A.E.; Sahai, I.; Bahena, P.; Reichert, S.L.; Loh, A.; et al. Recurrent de novo mutations affecting residue Arg138 of Pyrroline-5-carboxylate synthase cause a progeroid form of Autosomal-Dominant cutis laxa. Am. J. Hum. Genet. 2015, 97, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Shetty, R.; Sathyanarayanamoorthy, A.; Ramachandra, R.A.; Arora, V.; Ghosh, A.; Srivatsa, P.R.; Pahuja, N.; Nuijts, R.M.; Sinha-Roy, A.; Mohan, R.R. Attenuation of lysyl oxidase and collagen gene expression in keratoconus patient corneal epithelium corresponds to disease severity. Mol. Vis. 2015, 21, 12–25. [Google Scholar] [PubMed]
- Mas Tur, V.; MacGregor, C.; Jayaswal, R.; O’Brart, D.; Maycock, N. A review of keratoconus: Diagnosis, pathophysiology, and genetics. Surv. Ophthalmol. 2017, 62, 770–783. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disorder | Collagen Type | Genetic Alteration b | Major Clinical Features b |
|---|---|---|---|
| Ehlers-Danlos syndrome (EDS) | COL1 COL3 COL5 | Mutations in more than a dozen genes have been found to cause EDS (#130000). The classical type 1 and 2 (#130010) result from mutations in either the COL5A1 (*120215) or COL5A2 (*120190) gene. Other genes involved are COL1A1 (*120150), COL1A2 (*120160), COL3A1 (*120180). Mutation in COL1A1 or COL1A2 lead to the deletion of exon 6 of the mRNA coding the α1 (EDS VIIA, #130060) or the α2 chain (EDS VIIB, #617821) of type I collagen, respectively. Inheritance is autosomal dominant. | EDS is the name associated with at least nine phenotypically characterized clinical entities, which result from different types of mutations in distinct collagen genes and several collagen processing genes. These disorders are biochemically and clinically distinct although they all manifest structural weakness in connective tissue as a result of defects in the structure and function of collagens [76]. Although all types of EDS affect joints and skin, additional features vary by type. Severity ranges from mild to severe. Joint hypermobility occurs with most forms of EDS. Infants with hypermobile joints often have weak muscle tone, which can delay the development of motor skills such as sitting, standing and walking. The loose joints are unstable and prone to dislocation and chronic pain. Many EDS patients have soft, velvety skin that is highly stretchy (skin hyperextensibility) and fragile. Affected individuals tend to bruise easily and in some cases, they show atrophic scars. People with the classical form of EDS experience wounds that split open with little bleeding and leave scars that widen over time to create characteristic “cigarette paper” scars. |
| Mutations in the COL3A1 have been identified in the vascular type of EDS (#130050). | The vascular type can involve serious and potentially life-threatening complications due to unpredictable tearing of blood vessels. This rupture can cause internal bleeding, stroke and shock. The EDS vascular type is also associated with an increased risk of organ rupture, including tearing of the intestine or the uterus (womb) during pregnancy. | ||
| Mutations in procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 gene (PLOD1, *153454) and FK506 binding protein 14 (FKBP14, *614505) are responsible for the EDS kyphoscoliotic type I and II (EDSKMH, #225400; #614557). Inheritance is autosomal dominant in both cases. PLOD1 catalyses the hydroxylation of lysyl residues in collagen-like peptides, which are critical for the stability of intermolecular crosslinks. FKBP14 acts at the level of the protein folding in the ER, including components of the ECM (COL1, COL3, COL6 and fibronectin). | In addition to the classical symptoms of EDS, patients with EDSKMH I and II are characterised also by progressive kyphoscoliosis with muscle hypotonia from birth, joint laxity, gross motor delay, severe skin hyperelasticity, easy bruising, fragility of sclerae, myopathy and hearing loss [77]. | ||
| The Musculocontractural Type I and type II form of EDS (EDSMC1, #601776; EDSMC2, #615539) are caused by recessive loss-of-function mutations in the carbohydrate sulfotransferase 14 (CHST14, *608429) and in the dermatan sulphate epimerase (DSE, *605942) genes, respectively. The genes encode enzymes involved in the dermatan sulphate (DS) bio-synthesis that is involved in the assembly of collagen fibril. Mutations in both genes lead to the intracellular retention of COL1 and COL3 and a reduced deposition of collagen types I, III, V and VI in the ECM. | EDSMC1 and 2 share most of the clinical features, even though the majority of cases (31) refer to the EDSMC1 and only three cases are reported for the EDSMC2 type [78]. The two syndromes are characterised by progressive kyphoscoliosis, adducted thumbs in infancy or clenched fists and talipes equinovarus, hands with atypically shallow palmar creases and tapering fingers, joint hypermobility, clubfoot, arachnodactyly, elastic skin and poor wound healing. Craniofacial features include brachycephaly, large fontanel, hypertelorism, downslanting palpebral fissures, microcorneae, strabismus, prominent nasolabial folds, short philtrum, thin upper lip, small mouth, high palate and microretrognathia. EDSMC neonates show distal arthrogryposis and muscular hypotonia [78,79]. | ||
| The Spondylodysplastic Type 1 (also known as progeroid form of EDS) and 2 forms of EDS (EDSSPD1, #130070; EDSSPD2, #615349) are caused by mutations in the β-1,4-Galactosyltransferase 7 gene (B4GALT7, *604327) and β-1,3-Galactosyltransferase 6 (B3GALT6, *615291), respectively. The genes encode enzymes involved in the production and proper folding of collagen in connective tissue. The Spondylocheiro dysplastic form of EDS (SCD-EDS) results from mutations in the membrane-bound zinc transporter SLC39A13 (*608735) and has a reliable clinical overlap with EDSSPD1-2. Mutations in SLC39A13 result in increased Zn2+ content inside the endoplasmic reticulum, which inhibits the proper collagen crosslinking and the stability of the collagen triple helix. EDSSPD1 is an autosomal dominant disease whereas EDSSPD2 and SCD-EDS have an autosomal recessive inheritance. | Patients with EDSSPD1-2 showed short stature, muscle hypotonia, radioulnar synostosis and mild to severe intellectual disability (ID). In addition, they present facial dysmorphism, hyperextensible skin, joint hypermobility (JHM), single transverse palmar crease, severe hypermetropia, limb bowing and osteopenia [80]. | ||
| The Dermatosparaxis type of EDS (EDSDERMS, #225410) results from mutations in disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS2,*604539), the gene encoding the procollagen peptidase that cleavages the N-propeptide of the fibrillar procollagens types I-III and V | The EDSDERMS is characterized by skin that sags and wrinkles. Extra (redundant) folds of skin may be present as affected children get older [81]. | ||
| Osteogenesis Imperfecta (OI) | COL1 | Mutations in the COL1A1 (*120150) and COL1A2 (*120160) genes are responsible for more than 90% of all cases of OI (#166200). Most of the mutations causative of OI type I affect COL1A1 gene and result in reduced levels of COL1, whereas those responsible for most of OI types II (#166210), III (#259420) and IV (#166220) cases occur in COL1A1 or COL1A2 genes and impair COL1 structure. The inheritance is autosomal dominant. | At least four biochemically and clinically distinguishable forms of OI have been identified associated to defects in COL1. These are named as OI type I (mild), type II (perinatal lethal), type III (deforming) and type IV (mild deforming). A defect in COL1 structure weakens connective tissues, particularly bones. All four forms of OI present reduced levels of COL1 and brittle bones that break easily. Multiple fractures result in bone deformities. Additional symptoms may include blue sclera, short height, loose joints, hearing loss, breathing and teeth problems, cervical artery dissection and aortic dissection [82,83]. |
| Mutations in Cartilage associated protein CRTAP, *605497), Prolyl 3-hydroxylase (P3H1, *610339) and Peptidyl-prolyl isomerase B (PPIB, *123841) genes results in OI type VII (#610682), VIII (#610915) and IX (#259440), respectively. CRTAP encodes a cartilage-associated protein whereas P3H1 an enzyme belonging to the collagen propyl hydroxylase family. PPIB encodes for a cyclophilins (Cyps) protein that catalyses the cis–trans isomerisation of peptide bonds. All these proteins are required for proper collagen synthesis, assembly and secretion. In these cases, the inheritance is autosomal recessive. | In addition to the four forms of OI previously described, eleven additional phenotypically related disorders in the OI family exist, all associated with bone fragility and low bone mass. Among the OI associated to collagen alterations, the type VII (mutations in CRTAP) is sometimes considered a lethal form with multiple fractures, long bone deformities, scoliosis and short stature [84]. The type VIII form of OI (mutations in P3H1) includes severe growth defects, skeletal demineralization, scoliosis, round face and proptosis [85]. The type IX (mutations in PPIB) is a very severe form of OI. Embryos dye during pregnancy or few months after birth. Radiographs and an autopsy showed the presence of shortened, bowed and fractured long bones without evident rhizomelia [86]. | ||
| The Serpin family H member 1 (SERPINH1, *600943) gene encodes a collagen-binding protein that has chaperone activity in the endoplasmic reticulum. Mutations in SERPINH1 cause the type X OI (#613848), whose inheritance is autosomal recessive. | Types X OI is a severe deforming form of the disorder characterized by aberrant collagen crosslinking, folding and chaperoning [87]. | ||
| Absence of FK506 binding protein 10 (FKBP10, *607063) in recessive type XI OI (#610968) leads to reduced collagen cross-linking and deposition. FKBP10 encodes a chaperone that contributes to type I procollagen folding. Mutations in this gene affect its secretion. | Clinical hallmarks of OI type XI are congenital contractures. All the others clinical data on the 29 patients with OI type XI (mutations in FKBP10) are limited and heterogeneous regarding the age of onset, the number of fractures, the type of affected bones and the severity of the disorder [88]. | ||
| The recessive form of OI type XIII (OI13, #614856) is caused by mutations in the BMP1 (*112264) gene, which is involved in the processing of the C-propeptides of procollagens types I-III and the proteolytic activation of the enzyme lysyl oxidase, necessary for the formation of covalent cross-links in collagen and elastic fibres. | The OI type XIII is characterized by normal teeth, faint blue sclerae, severe growth deficiency, borderline osteoporosis and an average of 10–15 fractures a year affecting both the upper and lower limbs and with severe bone deformity. | ||
| The Secreted protein acidic and cysteine rich (SPARC,*182120) gene encodes a glycoprotein that binds to COL1 and other ECM proteins. Mutations in this gene are responsible for the type XVII OI (#616507) and seem to result in the over-modification of collagen during triple-helical formation. The inheritance is autosomal recessive. | Two clinical cases have been reported: the first is a girl from North Africa with low bone mineral density (BMD), scoliosis, short stature, mild joint hyperlaxity, weak underdeveloped muscles of the lower extremities, bowing of both humeri and speech delay. The second patient is an Indian girl, who had a left hip dislocation at the age of 10 weeks, muscle hypotonia and gross motor developmental delay. Other features are decreased calf muscle mass, joint hyperlaxity and soft skin [89]. | ||
| Caffey disease, also called infantile cortical hyperostosis | COL1 | The COL1A1 (*120150) variant c.3040C>T (p.Arg836Cys) in exon 41 is the pathogenic variant currently identified in all individuals with Caffey disease (#114000). Inheritance is autosomal dominant but not all people who inherit the mutation develop signs and symptoms. The amino acid change leads to COL1 fibrils that are variable in size and shape. | Caffey disease is characterised by excessive new bone formation (hyperostosis) in early infants. Affected bones may double or triple in width and include jawbone, scapulae, clavicles and the shafts (diaphyses) of long bones in arms and leg. Affected babies are frequently feverish and irritable. They show swelling of joints, pain and redness of affected areas. Usually, there is spontaneous resolution of the inflammatory signs within few months or years. Rare cases of recurrence have also been described [90]. |
| Alpha-2-Deficient Collagen Disease | COL1 | In 1974 Meigel and co-authors [91] described a 10-year-old son of consanguineous parents, with an apparently ‘new’ connective tissue disorder. The clinical and radiologic abnormalities were reminiscent of both Marfan syndrome and osteogenesis imperfecta. Study of cultured fibroblasts showed a complete failure of synthesis of α-2 chains of collagen. | |
| Spondyloepiphysea l dysplasia congenita (SED) | COL2 | SED congenita (# 183900) is caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. The inheritance is autosomal dominant. | SED congenita is a chondrodysplasia characterized by short spine, barrel-shaped chest, abnormal epiphyses and flattened vertebral bodies. Skeletal features are manifested at birth and evolve with time. Other features include myopia and/or retinal degeneration with retinal detachment and cleft palate [92]. |
| Stanescu type of spondyloepiphyseal dysplasia (SEDSTN) | COL2 | SEDSTN (#616583) is caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. The inheritance is autosomal dominant. | Spondyloepiphyseal dysplasia with accumulation of glycoprotein in chondrocytes has been designated the “Stanescu type”. Clinical hallmarks include progressive joint contracture with premature degenerative joint disease, particularly in the knee, hip and finger joints and swollen interphalangeal joints of the hands. The affected individuals are not short, despite the presence of a short trunk. Radiologically, spondylar and epiphyseal abnormalities are quite conspicuous. Other clinical characteristics are generalized platyspondyly, hypoplastic pelvis, epiphyseal flattening with metaphyseal splaying of the long bones and enlarged phalangeal epimetaphyses of the hands [93,94]. |
| Multiple epiphyseal dysplasia with myopia and conductive deafness (EDMMD) | COL2 | EDMMD (#132450) is caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. The inheritance is autosomal dominant. | EDMMD is characterized by epiphyseal dysplasia associated with progressive myopia, retinal thinning, crenated cataracts, conductive deafness, joint pain, deformity, waddling gait and short stature. In 1978 Beighton and colleagues [95] described an Afrikaner family in South Africa in which the mother, two sons and one daughter had a syndrome of multiple epiphyseal dysplasia, myopia and conductive deafness. The patients had short stature, brachydactyly, genu valgus deformity and dysplasia of the epiphyses. The epiphyses around the knee joint were flattened, the femoral necks were widened and the vertebral bodies were mildly reduced in height and were concave on their upper and lower surfaces. |
| Achondrogenesis type II (ACG2) | COL2 | ACG2 (#200610) is caused by mutations in COL2A1 gene (*120140) on chromosome 12q13. The inheritance is autosomal dominant but somatic and germline mosaicism have also been reported [96]. | ACG2 is characterized by severe micromelic dwarfism with small chest and prominent abdomen. Other clinical features include incomplete bone ossification and disorganization of the costochondral junction. The cartilage appears as abnormal gelatinous texture and translucent [75]. |
| Czech dysplasia | COL2 | Czech dysplasia (#609162) is caused by heterozygous mutations in COL2A1 gene (*120140) on chromosome 12q13. The inheritance is autosomal dominant. | Czech dysplasia is a skeletal dysplasia characterized by early and progressive onset, shortening of the third and fourth toes caused by metatarsal hypoplasia [97]. Affected individuals have a normal stature but usually complain of severe joint pain before adolescence. Clinical signs are restricted mobility in the lower limb joints and kyphoscoliosis. Skeletal radiographs reveal signs of pseudorheumatoid. Narrow joint spaces and flattened epiphyses platyspondyly with irregular endplates and elongated vertebrae can be observed in the most severe cases. Only five affected families from the Czech Republic have been so far reported [98]. |
| Legg-Calve-Perthes disease (LCPD) | COL2 | LCPD (#150600) is caused by heterozygous mutation in the COL2A1 (*120140) gene on chromosome 12q13. The inheritance is autosomal dominant. | LCPD is a form of avascular necrosis of the femoral head (ANFH; #608805) that affects hip development in growing children. It is due to loss of circulation in the femoral head. Radiology does not permit an early diagnosis that depends on the phase of disease progression through ischemia, revascularization, fracture and collapse, repair and remodelling of the bone. LCPD affects more often boys who are usually shorter than their peers [99]. |
| Osteoarthritis with mild chondrodysplasia (OSCDP) | COL2 | OSCDP (#604864) is caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. The inheritance is autosomal dominant. | OSCDP is a common disease that produces joint pain and stiffness together with radiologic evidence of progressive degeneration of joint cartilage. Several cases have been reported, included family members over various generations [95,100,101]. Major features are primary osteoarthritis associated with mild chondrodysplasia. Over the years the range of motion becomes limited. In about 60% of affected persons, abnormalities of the vertebral bodies consistent with mild chondrodysplasia have been described, including platyspondyly, irregular end plates, herniations within the vertebral bodies (Schmorl nodes) and anterior wedging. Other minor changes include iliac exostoses. |
| Torrance type of platyspondylic lethal skeletal dysplasia (PLSD-T) | COL2 | PLSD-T (#151210) can be caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. The disease is transmitted in an autosomal dominant manner. All the patients analysed so far have mutations in the C-propeptide domain of COL2A1, which lead to altered biosynthesis. The phenotype could result from a combination of diminished collagen fibril formation, toxic effects through the accumulation of unfolded collagen chains inside the chondrocytes and/or alteration of a putative signalling function of the C-propeptide. | PLSD-T is a rare skeletal dysplasia characterized by platyspondyly, brachydactyly and metaphyseal changes. Radiology reveals decreased ossification of the skull base, short thin ribs, hypoplastic pelvis with wide sacrosciatic notches and flat acetabular roof, short tubular long bones with ragged metaphyses and bowing of the radius. Histologically, the growth plate appeared relatively normal. The resting cartilage appeared hypercellular with large chondrocytes [102,103]. Though generally lethal in the perinatal period, a few long-term survivors with PLSD-T have been reported [104]. Some patients also present shortening of long bones, degenerative changes in the proximal femora, limited elbow extension, midface hypoplasia, myopia, deafness and mental retardation [105]. |
| Strudwick type of spondyloepimeta-physeal dysplasia (SEMD) | COL2 | SEMD (#184250) is an autosomal dominant disorder caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. | SEMD clinical features include severe dwarfism, marked pectus carinatum and scoliosis. Cleft palate and retinal detachment are frequently associated. Distinctive radiographic feature is irregular sclerotic changes, described as “dappled” in the metaphyses of the long bones that are caused by alternating zones of osteopenia and osteosclerosis [106]. |
| Spondyloperipheral dysplasia | COL2 | Spondyloperipheral dysplasia (#271700) is autosomal dominant disorder caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. | The disorder is a skeletal dysplasia with platyspondyly and brachydactyly E-like changes (short meta-carpals and metatarsals, short distal phalanges in the hand and feet) [107]. |
| Stickler syndrome (STL) | COL2, COL9 COL11 | Pathogenic variants in one of six genes (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2 and COL9A3) can be associated with Stickler syndrome. STL is inherited in autosomal dominant manner when mutated in COL2A1, COL11A1 or COL11A2, in autosomal recessive manner when mutated in COL9A1, COL9A2, or COL9A3. | STL is a genetically heterogeneous connective tissue disorder characterized by myopia, cataract and retinal detachment, conductive and sensorineural hearing loss. Additional findings may include mid–facial underdevelopment and cleft palate, mild spondyloepiphyseal dysplasia and/or precocious arthritis. Variable phenotypic expression occurs within and among families. Interfamilial variability is partially explained by locus and allelic heterogeneity [108]. |
| Stickler syndrome type I (STL1) | STL1 (#108300), also called the membranous vitreous type, is caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. | STL1 patients usually display a congenital vitreous abnormality consisting of a vestigial gel in the retrolental space, bounded by a highly folded membrane. Most affected individuals are at high risk for retinal detachment. Systemic features typically seen in STL1 are premature osteoarthritis, cleft palate, hearing impairment and craniofacial abnormalities [109]. | |
| Stickler syndrome type II (STL2) | STL2 (#604841), sometimes called the beaded vitreous type, is caused by heterozygous mutation in COL11A1 gene (*120280) on chromosome 1p21. | Patients affected by STL2 are myopic, rarely with paravascular lattice retinopathy. They frequently present cataract or are aphakic or pseudophakic. Retinal detachment, either mono- or bi-lateral may appear in the 3rd decade. Moreover, COL11A1 mutations are associated by early-onset hearing loss [110]. | |
| Stickler syndrome type III (STL3) | STL3 (#184840) or “nonocular Stickler syndrome” has been recently reclassified as form of otospondylomegaepiphyseal dysplasia or Weissenbacher-Zweymuller syndrome (OSMEDA or WZS). It is caused by heterozygous mutations in COL11A2 gene (*120290) on 6p21 chromosome. | Patients affected by STL3 have typical facial features, including midface hypoplasia combined with hearing impairment. No ocular abnormalities are reported. They present relatively short extremities with abnormally large knees and elbows but normal total body length. Diagnostic radiologic findings are enlarged epiphyses combined with moderate platyspondyly, mainly in the lower thoracic region [111]. | |
| Stickler syndrome type IV (STL4) | STL4 (#614134) is caused by homozygous mutation in COL9A1 gene (*120210) on chromosome 6q13. | Individuals affected by STL4 have moderate-to-severe sensorineural hearing loss, moderate-to-high myopia with vitreoretinopathy, cataracts and epiphyseal dysplasia [112]. The vitreous abnormality resembles an aged vitreous rather than the typical membranous, beaded or non-fibrillar type. | |
| Stickler syndrome type V (STL5) | STL5 (#614284) is caused by homozygous mutation in COL9A2 gene (*120260) on chromosome 1p34. | One family with STL5 has been reported. Major clinical findings are high myopia, vitreoretinal degeneration, retinal detachment, hearing loss and short stature. None of the family members was known to have cleft palate and, although there was short stature in childhood, normal height was found in adults [108]. | |
| Stickler syndrome atypical | The atypical form of STL (#609508) with predominantly ocular findings is caused by mutation in COL2A1 gene (*120140). The inheritance is autosomal dominant. | Patients display high myopia and retinal detachment. Systemic features of premature osteoarthritis, cleft palate, hearing impairment and craniofacial abnormalities are very mild or absent [113]. | |
| Familial avascular necrosis of the femoral head-1 (ANFH1) | COL2 | ANFH1 (#608805) is an autosomal dominant disorder caused by heterozygous mutation in COL2A1 gene (*120140) on chromosome 12q13. | ANFH1 is a debilitating disease affecting young adults between 35 and 55 years of age. The disorder is characterized by progressive pain in the groin, mechanical failure of the subchondral bone and degeneration of the hip joint. Nearly half of patients require hip replacement before 40 years of age [114]. |
| Kniest dysplasia | COL2 | Kniest dysplasia (#156550) is caused by mutations in COL2A1 gene (*120140). The inheritance is autosomal dominant. | Patients have short stature, flat facial profile, high myopia, risk of retinal detachment, cleft palate, deafness, high risk of severe degenerative joint disease and odontoid hypoplasia leading to risk of atalantoaxial instability and paralysis. Other features include neonatal respiratory distress, infantile hypotonia, abnormal oval-shaped vertebra at birth and later platyspondyly, shortened, “dumbbellshaped” long bones, with splaying of the epiphyses and metaphyses [115]. |
| Alport syndrome | COL4 | Alport syndrome is a clinically and genetically heterogeneous nephropathy. Approximately 80% of cases are transmitted as an X-linked semi-dominant condition due to COL4A5 mutations. 20% of cases are autosomal recessive due to mutation in either COL4A3 or COL4A4. Same families with autosomal-dominant Alport syndrome have been reported, either caused by COL4A3 or COL4A4 mutations. | Alport syndrome is characterized by progressive nephritis associated with hearing loss and sometime ocular lesions. Patients experience progressive loss of kidney function. The majority of affected individuals have blood (haematuria) and high levels of proteins (proteinuria) in their urine, which indicate impaired kidney function. Many patients also develop hypertension and at end-stage renal disease. Ocular anomalies are frequent in Alport syndrome and they can precede proteinuria in 40% of patients. Anterior lenticonus, abnormal coloration of the retina, lens rupture, cataracts and corneal erosions can be found [116]. Pregnancy of patients with Alport syndrome is very challenging and often complicated by deterioration of renal function, preeclampsia, severe placental dysfunction and sometime acute renal failure. Preterm delivery is frequent [117]. |
| Alport syndrome autosomal dominant | The autosomal dominant form of Alport syndrome (#104200) is caused by heterozygous mutation in COL4A3 gene (*120070). | ||
| Alport syndrome X-LINKED (ATS) | ATS (#301050) is caused by mutations in COL4A5 (*303630) gene. The inheritance is dominant. | ATS males are more severely affected than females. Men have a 90% chance of developing end-stage kidney disease by age 40. Patients with large deletions or nonsense mutations have significantly earlier onset than those with missense mutations. The majority (95.5%) of women with COL4A5 mutations develop microscopic haematuria [118]. | |
| Leiomyomatosis, diffuse, with Alport syndrome (DL-ATS) | DL-ATS (#308940) is caused by large deletions involving COL4A5 (*303630) and COL4A6 (*303631) genes. Likely an X-linked semi-dominant inheritance. | DL-ATS reveals the Alport syndrome features associated with diffuse leiomyomatosis [119]. | |
| Alport syndrome autosomal recessive | This form of Alport syndrome (#203780) is caused by mutations in COL4A3 (*120070) or COL4A4 (*120131) gene. | Autosomal recessive Alport syndrome presents as gross proteinuria in childhood and progression to end-stage kidney disease often before the fourth decade [120]. | |
| Autosomal dominant mental retardation-34 (MRD34) | COL4 | MRD34 (#616351) is caused by heterozygous mutation in COL4A3BP (*604677) gene on chromosome 5q13. The inheritance is autosomal dominant. | Patients with MRD34 present unremarkable perinatal history and delivery with a normal birth weight. Neonatal feeding difficulties may occur. Psychomotor development is delayed and speech skills limited. Auto-mutilation behaviour and anxiety are observed. Normal growth parameters and no evident dysmorphism are recorded in adults [121,122]. |
| Retinal arterial tortuosity (RATOR) | COL4 | RATOR (#180000) is caused by heterozygous mutation in COL4A1 gene (*120130) on chromosome 13q34. The inheritance is autosomal dominant. One single family with approximately 20 familial cases has been reported so far. | RATOR is an uncommon condition characterized by marked tortuosity of second- and third-order retinal arteries with normal first-order arteries and venous system. Typically, the vascular tortuosity is predominantly located at the macular and peripapillary area and develops during childhood or early adulthood. Although the disease may be asymptomatic, most patients complain of variable degrees of transient vision loss due to retinal haemorrhage following physical exertion or minor trauma. Involvement of non-ocular vascular beds has not been demonstrated in most cases but occasionally other associated vascular abnormalities have been recorded, including malformations in the Kieselbach nasal septum, spinal cord vascular mass, telangiectasis of bulbar conjunctiva and internal carotid artery aneurysm [123]. |
| Hereditary angiopathy with nephropathy, aneurysms and muscle cramps (HANAC) | COL4 | HANAC (#611773) is caused by heterozygous mutation in COL4A1 gene (*120130) on chromosome 13q34. The inheritance is autosomal dominant. | HANAC syndrome is characterized by angiopathy that affects several parts of the body. Patients present kidney alterations consisting of multiple renal cysts and sometimes haematuria. The brain is only mildly affected and intracranial aneurysms causing haemorrhagic stroke can occur. Leukoencephalopathy is found in about half of affected individuals whereas muscle cramps are experienced by most of patients in early childhood. In addition, patients may manifest eye problems, like arterial retinal tortuosity, cataract and abnormality called Axenfeld-Rieger anomaly [124]. |
| Small vessel disease of the brain with or without ocular anomalies (BSVD) | COL4 | BSVD (#607595) is caused by heterozygous mutation in COL4A1 gene (*120130) on chromosome 13q34. The inheritance is autosomal dominant. | BSVD is characterized by a wide spectrum of symptoms of varying severity including porencephaly variably associated with eye defects (retinal arterial tortuosity, Axenfeld-Rieger anomaly, cataract) and systemic findings such as kidney involvement, muscle cramps, cerebral aneurysms, Raynaud phenomenon, cardiac arrhythmia and haemolytic anaemia. Stroke is often the first symptom and is usually caused by haemorrhagic rather than ischemic stroke. Patients also have leukoencephalopathy and may experience infantile hemiparesis, seizures and migraine headaches accompanied by visual auras [125]. |
| Porencephaly | COL4 | Porencephaly is an autosomal dominant disorder characterize by mutations in COL4A1 (*120130) or COL4A2 (*120090) genes on chromosome 13q34. | It is a neurological disorder characterized by fluid-filled cysts or cavities in the brain and is thought to result from disturbed vascular supply leading to cerebral degeneration. Affected individuals have delayed growth and development, hypotonia, spastic hemiplegia, seizures, migraine headaches, speech problems and intellectual disability with variable severity [126]. |
| Porencephaly-1 (POREN1) | POREN1 (#175780) is caused by mutations in COL4A1 gene. | POREN1 is more common. It is usually unilateral and results from destructive lesions. | |
| Porencephaly-2 (POREN2) | POREN2 (#614483) is caused by mutations in COL4A2 gene. | POREN2 is usually symmetrical and results from developmental malformation. | |
| Schizencephaly | COL4 | Some patients with schizencephaly (#269160) have mutations in COL4A1 (*120130) gene. | Schizencephaly is a very rare cortical malformation that results in grey matter line clefts impacting one or both sides of the brain. Two types of schizencephaly have been described, depending on the size of the area involved and on the separation of the cleft lips. The clinical picture is mainly based on the presence of motor deficits and mental retardation but the severity of the symptoms varies depending on the size and location of the clefts and on the presence of associated cerebral malformations. Patients with type I are almost normal, they may have seizures or motor impairment. Type II is associated with mental retardation, seizures, hypotonia, spasticity, inability to walk or speak and blindness [127]. |
| Susceptibility to intracerebral haemorrhage (ICH) | COL4 | ICH (#614519) may be due to mutations in COL4A2 (*120090) or COL4A1 (*120130) genes on chromosome 13q34. The inheritance is autosomal dominant. | Few patients with adult-onset haemorrhagic stroke have been reported. The mutated vascular collagen diminishes the tensile strength of vessels and increases their fragility, which can lead to haemorrhage [128]. |
| X-linked deafness-6 (DFNX6) | COL4 | DFNX6 (#300914) is caused by mutation in COL4A6 gene (*303631) on chromosome Xq22. One family has been reported so far. | The symptoms vary in male and female patients affected by this disorder. The severe bilateral sensorineural hearing loss apparent in infancy affects only males, who present bilateral malformation of the cochlea with incomplete separation from the internal auditory canal. Language skills in these patients are severely restricted. Female patients develop mild to moderate hearing impairment in the third/fourth decades of life and rarely hearing loss in the first decade of life [129]. |
| Benign familial haematuria (BFH) | COL4 | BFH (#141200) are caused by mutations in COL4A3 (*120070) or COL4A4 (*120131) gene, both of which map on chromosome 2q36. The inheritance is autosomal dominant. | BFH is characterized by the presence of persistent or recurrent haematuria, usually detected in childhood. Haematuria remains isolated and never results in end-stage renal disease. Diffuse attenuation of the glomerular basement membrane is usually considered the hallmark of the condition but it is not specific [130]. |
| Bethlem myopathy-1 (BTHLM1) | COL6 | BTHLM1 (#158810) is caused by mutations in COL6A1 (*120220), COL6A2 (*120240) or COL6A3 (*120250) genes, giving rise to the altered or even lack of type VI collagen. Both recessive and dominant mutations have been reported. | The disease is characterized by progressive muscle weakness and joint stiffness (contractures). The features can appear at any age, in some cases before birth (decreased foetal movements) in other cases during infancy with joint laxity (loose joints) and hypotonia (weak muscle tone). Later, during childhood, patients develop contractures in their fingers, wrists, elbows and ankles. When adult, they may develop weakness in respiratory muscles, which result in breathing difficulty. The mild form may also reveal skin abnormalities, including follicular hyperkeratosis on the arms and legs; soft, velvety skin on the hand palms and feet soles; abnormal wound healing resulting in shallow scars [131]. |
| Ullrich congenital muscular dystrophy-1 (UCMD1) | COL6 | UCMD1 (#254090) is caused by mutations in COL6A1 (*120220), COL6A2 (*120240) or COL6A3 (*120250) genes, giving rise to the altered or even lack of type VI collagen. The disease is transmitted in an autosomal recessive manner and only in rare cases in a dominant pattern. | Patients suffer from a severe muscle weakness beginning soon after birth. Some affected individuals are never able to walk and others can walk only with support. Several lose ambulation ability in adolescence. Progressive scoliosis and deterioration of respiratory function is a typical feature. Some patients need continuous mechanical ventilation to help them breathing. Affected individuals develop contractures in their neck, hips and knees, which further impair movement. There may be joint laxity in patient fingers, wrists, toes, ankles and other joints. As in BTHLM1, some people with UCMD1 have follicular hyperkeratosis [132]. |
| Autosomal recessive myosclerosis | COL6 | The autosomal recessive myosclerosis (#255600) has an autosomal recessive inheritance and is caused by mutations in COL6A2 gene (*120240). One family has been reported so far. | The disorder is characterized by chronic inflammation of skeletal muscle with hyperplasia of the interstitial connective tissue. The clinical symptoms include slender muscles with “woody” consistency and restriction of movement of many joints because of muscle contractures. Muscles are thin and may result sclerotic on palpation. The few patients so far described showed difficulty in running and climbing stairs and had Achilles tendon contractures during early childhood. Skeletal muscle biopsies showed a myopathic pattern with fibrosis, proliferation of endomysial and perimysial connective tissue, variation of myofibre diameter. Increased serum creatine kinase was also found [133]. |
| Dystonia 27 (DYT27) | COL6 | DYT27 (#616411) is caused by compound heterozygous mutations in COL6A3 gene (*120250) on chromosome 2q37. It is an autosomal recessive disorder. | Neurological disorder characterized by the onset of segmental isolated dystonia involving the face, neck, bulbar muscles and upper limbs in the first two decades of life. Few cases have been reported and the symptoms included dystonic action and postural tremor, writer’s cramp, oromandibular and laryngeal dystonia [134]. |
| The dystrophic forms of epidermolysis bullosa (DEB) | COL7 COL17 | The autosomal dominant form of epidermolysis bullosa dystrophica (DDEB, #131750) is caused by heterozygous mutations in COL7A1 gene (*120120) on chromosome 3p21. The autosomal recessive dystrophic form of epidermolysis bullosa (RDEB, #226600) and the RDEB localized variant (#226650) are caused by homozygous or compound heterozygous mutations in COL17A1 gene (*113811). | Epidermolysis bullosa (EB) is a term referring to a family of disorders that are associated with excessive blistering in response to mechanical injury or trauma. Microscopic examination of the skin shows cleavage below the basement membrane within the papillary dermis. The signs and symptoms of this condition vary widely among affected individuals. In mild cases, blistering may primarily affect the hands, feet, knees and elbows. Severe cases involve widespread blistering leading to vision loss, disfigurement and other serious medical problems such as strictures of the gastrointestinal tract leading to poor nutrition. Patients show an increased risk of developing aggressive squamous cell carcinoma. Kids with EB are often defined “butterfly wing” children because of their extremely fragile skin, which can shed at the slightest touch. DEB is one of the major forms of EB. DDEB and RDEB are also known as Cockayne-Touraine disease and Hallopeau-Siemens disease, respectively [135]. Variations in severity are observed among the different forms of RDEB. Notably, a functional SNP in MMP1 (*120353) promoter is associated with high severity in RDEB. Since COL7 is degraded by MMP1, an imbalance between COL7 synthesis and degradation could worsen the RDEB phenotype [136]. |
| Nonsyndromic congenital nail disorder-8 (NDNC8) | COL7 | NDNC8 (#607523) is caused by heterozygous mutations in COL7A1 gene (*120120) on chromosome 3p21.1. The disorder is inherited in an autosomal dominant manner. | This form of isolated toenail dystrophy has been found in few Japanese families in which other members had the autosomal recessive dystrophic epidermolysis bullosa (RDEB, #226600) or the transient bullous dermolysis of the newborn (#131705), the features of which include dystrophic nails. The nail plates of the toes were buried in the nail bed and the free edge of the toenail was deformed and narrow [137]. |
| Fuchs endothelial corneal dystrophy-1 (FECD1) | COL8 | FECD1 (#136800) is caused by heterozygous mutations in COL8A2 gene (*120252) on chromosome 1p34. It is an autosomal dominant disorder. | FECD is a progressive, bilateral condition leading to reduced vision quality due to dysfunction of the corneal endothelial cells, a thin layer of cells in the back of the cornea that regulates the amount of fluid inside the cornea. FECD occurs when the endothelial cells die and the cornea becomes swollen with too much fluid. Corneal endothelial cells continue to die over time, resulting in further vision problems. Ultrastructural features include loss and attenuation of endothelial cells with thickening and excrescences (guttae) of the underlying basement membrane that are the clinical hallmark of FECD and that worsen with disease progression. As the endothelial layer develops confluent guttae in the central cornea, the cornea becomes dehydrated and clear [138]. In the USA about 5% of the over 40 population is affected by FECD and some early-onset cases are due to COL8A2 mutations. |
| Posterior polymorphous corneal dystrophy (PPCD2) | COL8 | A single family with PPCD2 (#609140) caused by heterozygous missense mutation in COL8A2 gene (*120252) has been described. Another family with one PPCD2 patient and few FECD cases, due to heterozygous missense mutation in COL8A2, has been described [139]. | Father and daughter with PPCD2 have been reported. The patients show a bilateral penetrating keratoplasty at the age of twenties (daughter) and fifties (father). The authors suggested that the underlying pathogenesis of FECD and PPCD2 may be related to disturbance of the role of COL8 in influencing the terminal differentiation of the neural crest-derived corneal endothelial cell [140]. |
| Multiple epiphyseal dysplasia (EDM) | COL9 | There are two types of EDM, which can be distinguished by their pattern of inheritance, the dominant and recessive types. EDM caused by mutations affecting collagen structures have an autosomal dominant transmission. Mutations in COL9A1, COL9A2 or COL9A3 genes are found in less than 5% of individuals with dominant EDM. | EDM is a clinically and genetically heterogeneous skeletal disorder, which is characterized by joint pain and stiffness, mild short stature and degenerative joint features. Both cartilage and bone development are affected, mainly at the ends of the long bones in the arms and legs (epiphyses). It has been suggested that mutations in COL9A1, COL9A2 or COL9A3 genes may cause COL9 to accumulate inside the cell or interact abnormally with other cartilage components. |
| Multiple epiphyseal dysplasia-2 (EDM2) | EDM2 (#600204) is caused by heterozygous mutation in COL9A2 gene (*120260) on chromosome 1p34. | EDM2 onset is usually in childhood, around 3-4 years of age and clinical variability is observed even within the same family [141]. | |
| Multiple epiphyseal dysplasia-3 (EDM3) | EDM3 (#600969) is caused by heterozygous mutation in COL9A3 gene (*120270). | EDM3 patients show early-onset short stature, waddling gait and pain/stiffness in the knees. Few patients experience involvement of elbow, wrist or ankle [142]. | |
| Multiple epiphyseal dysplasia-6 (EDM6) | EDM6 (#614135) is caused by heterozygous mutation in COL9A1 gene (*120210) on chromosome 6p13. One single family has been reported. | A 30-year-old proband was reported with knee pains and difficulty walking since 10 years of age. Radiographs showed early osteoarthritis of one knee, Schmorl nodes, endplate irregularities, anterior osteophytes in the thoracolumbar vertebrae and normal hips. The mother had the same mutation but she did not reveal any symptom before age 45 years [143] | |
| Schmid-type metaphyseal chondrodysplasia (MCDS) | COL10 | MCDS (#156500) is caused by heterozygous mutation in COL10A1 (*120110) gene on chromosome 6q22. MCDS is transmitted as an autosomal dominant trait. | MCDS is a rare genetic disorder characterized by short stature, short arms and legs (short-limbed dwarfism) and bowing of the long bones. Radiographic features include widening and irregularity of the growth plates, especially in the distal and proximal femora. These defects give rise to unusual “waddling” walk (gait) [144]. |
| Marshall syndrome (MRSHS) | COL11 | MRSHS (#154780) is an autosomal dominant genetic disorder caused by mutations in COL11A1 gene (*120280) on chromosome 1p21. | Patients have a distinctive flat midface with a flattened nasal bridge (saddle nose), nostrils that turn upward, widely spaced eyes, high myopia, cataracts and sensorineural hearing loss. Other symptoms include crossed eyes (esotropia), retinal detachment, glaucoma, protruding upper incisors (teeth) and a small or missing nasal bone [145]. |
| Fibrochondrogenesis-1 (FBCG1) | COL11 | FBCG1 (#228520) is a severe, autosomal recessive disorder caused by mutations in COL11A1 gene (*120280) on chromosome 1p21. | FBCG1 and FBCG2 are short-limbed skeletal dysplasia frequently lethal. The disorder is named for the disorganized cartilage growth plate in which chondrocytes have a fibroblastic appearance and the presence of fibrous cartilage extracellular matrix. Patients are characterized by short stature (dwarfism) and skeletal abnormalities. Affected individuals have shortened long bones in the arms and legs that are unusually wide at the ends (described as dumbbell-shaped). Hands and feet are relatively normal. Vertebrae are flattened (platyspondyly) and have a characteristic pinched or pear shape that is noticeable on x-rays. Ribs are typically short and wide and have metaphyseal cupping at both ends. Affected infants have a very narrow chest, which prevents the lungs from developing normally. Most infants are stillborn or die shortly after birth from respiratory failure. Some affected individuals have lived into childhood. Affected individuals who survive the neonatal period have high myopia, mild to moderate hearing loss and severe skeletal dysplasia [146]. |
| Fibrochondrogenesis-2 (FBCG2) | FBCG2 (#614524) can have an autosomal recessive or dominant inheritance due to mutations in COL11A2 gene (*120290) on chromosome 6p21.3. | ||
| Autosomal dominant deafness-13 (DFNA13) | COL11 | DFNA13 (#601868) is an autosomal dominant disorder caused by heterozygous mutation in COL11A2 gene (*120290) on chromosome 6p21. | A single family has been described, characterized by a dominant nonsyndromic postlingual hearing loss. The affected individuals experienced progressive hearing loss beginning in the second to fourth decades [147]. |
| Otospondylo-megaepiphyseal dysplasia, autosomal dominant (OSMEDA) | COL11 | The autosomal dominant OSMEDA (#184840), also known as Weissenbacher-Zweymuller syndrome (WZS), is caused by heterozygous mutation in COL11A2 gene (*120290) on chromosome 6p21. The disorder has an autosomal dominant transmission. | OSMED is characterized by skeletal abnormalities, distinctive facial features and severe hearing loss. The term “otospondylomegaepiphyseal” refers to the parts of the body that are affected: ears (oto-), bones of the spine (spondylo-) and the ends (epiphyses) of long bones in the arms and legs. The disorder is characterized by sensorineural hearing loss, relatively short extremities with abnormally large knees and elbows (enlarged epiphyses), vertebral body anomalies and characteristic facies. The diagnostic radiologic findings are enlarged epiphyses combined with moderate platyspondyly, mainly in the lower thoracic region. No ocular abnormalities are reported. Patients have typical facial features, including midface hypoplasia [148]. |
| Autosomal recessive (OSMEDB) | The autosomal recessive OSMEDB (#215150) is also caused by mutation in the COL11A2 gene. | ||
| Congenital myasthenic syndrome type 19 (CMS19) | COL13 | CMS19 (#616720) is an autosomal recessive disorder resulting from mutations in COL13A1 gene (*120350) on chromosome 10q22. | The congenital myasthenic syndromes (CMSs) are a heterogeneous group of inherited disorders resulting from impaired neuromuscular transmission and caused by mutations in genes involved in the formation or integrity of neuromuscular junctions (NMJs). CMS19 result in generalized muscle weakness, exercise intolerance and respiratory insufficiency. Patients present hypotonia, feeding difficulties and respiratory problems soon after birth. The severity of the weakness and disease course is variable [149]. |
| Epithelial recurrent erosion dystrophy (ERED) | COL17 | ERED (#122400) is caused by heterozygous mutation in COL17A1 gene (*113811) on chromosome 10q24. The disorder is transmitted as an autosomal dominant trait. | ERED is characterized by bilaterally painful recurrent corneal erosions. Erosions often are precipitated by relatively minor trauma and are often difficult to treat, lasting for up to a week. Fortunately, the erosions become less frequent as patients age and may cease altogether by the fifth decade of life. The onset is in the first decade of life (even in the first year of life) often with some subepithelial haze or blebs while denser centrally located opacities develop with time. Small grey anterior stromal flecks associated with larger focal grey-white disc-shaped, circular or wreath-like lesions with central clarity, in the Bowman layer and immediately subjacent anterior stroma, varying from 0.2 to 1.5 mm in diameter, may be diagnostic of ERED [150]. |
| Knobloch syndrome-1 (KNO1) | COL18 | KNO1 (#267750) is a hereditary autosomal recessive disorder caused by mutations in COL18A1 gene (*120328) on chromosome 21q22.3. | KNO1 is primarily characterized by severe vision problems and skull defects. Eye abnormalities include high myopia, cataracts, dislocated lens, vitreoretinal degeneration and retinal detachment. Skull defects range from occipital encephalocele to occult cutis aplasia [151]. |
| Congenital fibrosis of extraocular muscles-5 (CFEOM5) | COL25 | CFEOM5 (#616219) has an autosomal recessive inheritance and is caused by mutations in COL25A1 gene (*610004) on chromosome 4q25. A single family had been reported so far. | CFEOM include several different inherited strabismus syndromes characterized by congenital restrictive ophthalmoplegia affecting extraocular muscles innervated by the oculomotor and/or trochlear nerves. CFEOM5 has been reported in a single family with 3 sibs showing a congenital cranial dysinnervation affecting the ocular muscles. The patients had variable abnormal ocular motility without other systemic defects. Two sibs showed congenital ptosis with levator palpebrae muscle dysinnervation of one or both orbits. The levator palpebrae muscle was normally innervated by cranial nerve III (oculomotor nerve). The third sib had no ptosis but showed bilateral Duane retraction syndrome, exotropic in the right eye and esotropic in the left [152]. |
| Steel syndrome (STLS) | COL27 | STLS (#615155) displays an autosomal recessive inheritance due to mutations in COL27A1 gene (*608461) on 9q32 chromosome. Few cases have been reported who belong to the same family. | Patients affected by STLS present a characteristic facies, dislocated hips and radial heads, carpal coalition (fusion of carpal bones), short stature, scoliosis and cervical spine anomalies. The dislocated hips are resistant to surgical intervention [153]. |
| Disorder | Genetic Alteration | Link with the Disorder | Major Clinical Features a |
|---|---|---|---|
| Autosomal recessive dystrophic epidermolysis bullosa (RDEB) | A defect in collagenase MMP1 (*120353) has been implicated in RDEB (#226600). An association between disease severity and specific SNP in MMP1 gene (*120353) was found in three affected members of one family and in a cohort of 31 unrelated French RDEB patients [136]. The SNP results in increased transcript and active MMP1 protein levels. | COL7 is susceptible to degradation by the collagenase matrix metalloproteinases-1 (MMP1). An imbalance between COL7 synthesis and degradation could conceivably worsen the RDEB phenotype. | Patients with RDEB present generalized blisters at birth that result in extensive scarring and pseudosyndactyly. After birth, extensive blisters may affect the mucous membranes particularly the oral cavity, oesophagus and anal canal. Caused by chronic blood loss, inflammation, infection and poor nutrition, patients develop anaemia, failure to thrive, delayed puberty and osteoporosis. Patients usually do not survive more than 30 years due to severe renal complications or aggressive squamous cell carcinoma arising in the areas of repeated scarring [154]. |
| Aneurysm, abdominal aortic (AAA) | Mapped loci for AAA (#100070) include AAA1 (*100070) on chromosome 19q13, AAA2 (*609782) on chromosome 4q31, AAA3 (*611891) on chromosome 9p21 and AAA4 (*614375) on chromosome 12q13. Inheritance is autosomal dominant. | Several studies pointed to a role of MMPs in the end-stage of AAA. MMPs are enzymes capable of degrading connective tissue that may affect arterial walls by degrading collagens and other ECM components. Polymorphisms in MMP2, MMP3, MMP9 and MMP13 genes result in increased protein levels significantly associated to AAA risk. | AAA is characterized by chronic inflammation and ECM degradation of the aortic wall. The main symptoms of this condition are dysphasia, frontotemporal cerebral atrophy and frontotemporal dementia, speech disorder, memory impairment [155]. |
| Trichothiodystrophy 1, photosensitive form (TTD1) | TTD1 (#601675) is caused by homozygous or compound heterozygous mutation in the ERCC2/XPD gene (*126340) on chromosome 19q13. The gene encodes a helicase subunit of the transcription/repair factor TFIIH. The inheritance is autosomal recessive. | A reduced expression of COL6A1 (*120220), an abundant collagen of skin and connective tissue, has been shown in the skin of TTD patients with mutations in the ERCC2/XPD gene [156]. It has been shown that specific transcription deregulations in the cells of TTD patients with mutations in the ERCC2/XPD gene result in the overexpression of MMP1 gene. This event leads to hyper-secretion of active MMP1 enzyme and degradation of collagen type I in the dermis of TTD patient skin [157]. | TTD is characterized by hair abnormalities, physical and mental retardation, ichthyosis, signs of premature aging and cutaneous photosensitivity. The clinical spectrum of TTD varies widely from patients with only brittle, fragile hair to patients with the most severe neuroectodermal symptoms. TTD patients present sulphur-deficient brittle hair with a diagnostic alternating light and dark banding pattern (called ‘tiger’ tail banding) under polarizing microscopy. Common additional clinical features include collodion baby, characteristic facies, ocular abnormalities, short stature, decreased fertility and recurrent infections. TTD patients present a 20-fold higher mortality compared to the US general population [158,159]. |
| Atopic dermatitis (ATOD) | ATOD (#603165) is caused by the presence of a specific SNP (rs4688761) in COL29A1 gene (*611916), which encodes a novel epidermal collagen. The gene is on chromosome 3q22.1. Inheritance is autosomal dominant. | COL29A1 shows a specific gene expression pattern with the highest transcript levels in skin, lung and gastrointestinal tract, which are the major sites of allergic disease manifestation. Lack of COL29A1 expression in the outer layers of the epidermis of ATOD patients points to a role of collagen XXIX in epidermal integrity, whose breakdown is a clinical hallmark of AD [160]. | ATOD is a chronic inflammatory skin disease characterized by intensely itchy skin lesions. The onset of disease is typically observed during the first two years of life [161]. The hallmarks of atopic dermatitis are a chronic relapsing form of skin inflammation, a disturbance of epidermal barrier function that culminates in dry skin and IgE-mediated sensitization to food and environmental allergens. |
| Bruck syndrome (BRKS) | BRKS is a very rare autosomal recessive syndrome. Two forms are found: BRKS1 (#259450) is caused by mutations in FKBP10 (*607067) gene whereas BRKS2 (#609220) by mutations in PLOD2 (*601865) gene. | BRKS is characterized by bone fragility associated with congenital joint contractures. Patients commonly show short stature, skull wormian bones and kyphoscoliosis. Most cases had normal teeth, white sclera, normal cognitive functions and normal hearing. A few cases had dysmorphic features including triangular face and brachycephaly [162]. | |
| Bruck syndrome 1 (BRKS1) | BRKS1 (#259450) is caused by homozygous mutations in FKBP10 gene (*607063) on chromosome 17q21 resulting in FKBP65 loss of function. Inheritance is autosomal recessive. | Mutations in FKBP10 result in delay of type 1 procollagen secretion, incomplete stabilization of collagen trimer, reduced hydroxylation of the telopeptide lysyl residue (involved in intermolecular collagen cross-linking). | BRKS1 patients have short stature, high incidence of joint contractures, frequent fractures and scoliosis. |
| Bruck syndrome 2 (BRKS2) | BRKS2 (#609220) is caused by homozygous mutation in PLOD2 gene (*601865) on chromosome 3q24. Inheritance is autosomal recessive. | PLOD2 encodes the telopeptide lysyl hydroxylase required for the triple-helical cross-linking of collagen molecules. Mutations in this gene affect the instalment and secretion of collagen fibres from osteoblasts [163]. | No phenotypic differences between BRKS1 and BRKS2 have been reported. |
| Ehlers-Danlos syndrome (EDS) subtypes | The EDS subtypes are due to mutations in several genes, including PLOD1, FKBP14, ADAMTS2, ZNF469 and PRDM5. | ||
| EDS Kyphoscoliotic Type 1 (EDSKSCL1) | EDSKSCL1 (#225400) previously designated EDS6, is caused by homozygous or compound heterozygous mutation in the PLOD1 (*153454) gene on chromosome 1p36. Inheritance is autosomal recessive. | PLOD1 encodes a lysyl hydroxylase that catalyses the hydroxylation of lysine residues in X-lys-gly sequences of collagens and other proteins with collagen-like domains. This hydroxylation is essential for the stability of intermolecular collagen crosslinks. | EDSKSCL1 is characterized by skin fragility (easy bruising, friable skin, poor wound healing, widened atrophic scarring), scleral and ocular fragility/rupture, microcornea, facial dysmorphology. General features also include congenital muscle hypotonia, congenital or early onset kyphoscoliosis, joint hypermobility with subluxations or dislocations of shoulders, hips and knees [164]. |
| EDS Kyphoscoliotic Type, 2 (EDSKSCL2) | EDSKSCL2 (#614557) is caused by homozygous or compound heterozygous mutations in FKBP14 gene (*614505) on chromosome 7p15. Inheritance is autosomal recessive. | FKBP14 is an ER-resident protein belonging to the family of FK506-binding peptidyl-prolyl cis–trans isomerases (PPIases). It catalyses the folding of COL3 and interacts with COL3, COL4 and COLX [165]. | EDSKSCL2 is characterised by congenital hearing impairment (sensorineural, conductive, or mixed), follicular hyperkeratosis, muscle atrophy, bladder diverticula. |
| EDS dermatosparaxis Type (EDSDERMS) | EDSDERMS (#225410) is caused by mutation in ADAMTS2 (*604539) gene on chromosome 5q35. Inheritance is autosomal recessive. | ADAMTS2 encodes a procollagen protease that takes part to the processing of type I procollagen. | Dermatosparaxis means ‘tearing of skin.’ Patients present extreme skin laxity and fragility, easy bruising, extensive scar formation and joint laxity. Blue sclerae, micrognathia, umbilical hernia and postnatal growth retardation are reported [164]. |
| Brittle Cornea Syndrome1 (BCS1) | BCS1 (#229200) can be caused by homozygous mutation in the ZNF469 gene (*612078) on chromosome 16q24. Inheritance is autosomal recessive. | ZNF469 encodes a zinc-finger protein that likely acts as a transcription factor or extra-nuclear regulator factor for the synthesis or organization of collagen fibres. | BCS1 and BCS2 are associated with retinal microvascular abnormalities, keratoconus or keratoglobus, blue sclerae, extreme corneal thinning and a high risk of corneal rupture. Hyperelasticity of the skin without excessive fragility and hypermobility of the joints are other hallmarks of the disease [164]. |
| Brittle Cornea Syndrome2 (BCS2) | BCS2 (#614170) is caused by mutation in PRDM5 gene (*614161) on chromosome 4q27. Inheritance is autosomal recessive. | PRDM5 seems to regulate the expression of proteins involved in extracellular matrix development and maintenance, including COL4A1 and COL11A1. | BCS2 features overlap with BCS1. Systemic abnormalities included increased skin laxity, pectus excavatum, scoliosis, congenital hip dislocation, recurrent shoulder dislocation, high-frequency hearing loss, high-arched palate and mitral valve prolapse [166]. |
| CUTIS LAXA | Cutis laxa can be caused by mutations in either PYCR1 (*179035) or ALDH18A1 (#614438) gene. | Cutis laxa is a rare skin disorder characterized by wrinkled, redundant, inelastic and sagging skin due to defective synthesis of elastic fibres and other proteins of the ECM [167]. | |
| Cutis Laxa, autosomal recessive Type IIB (ARCL2B) | ARCL2B (#612940) is caused by homozygous or compound heterozygous mutation in the PYCR1 gene (*179035) on chromosome 17q25.3. Inheritance is autosomal recessive. | PYCR1 encodes the enzyme pyrroline-5-carboxylate reductase1, which catalyses the last step of proline synthesis. PYCR1 deficiency can affect the proper collagen formation. | ARCL2 is a more benign form of cutis laxa present at birth. Growth and developmental delay and skeletal anomalies are reported. Intellectual deficit and seizures have been reported in older patients [167]. Systemic manifestations are mild whereas pulmonary emphysema and cardiac anomalies are rare. |
| Cutis Laxa, autosomal recessive Type IIIB (ARCL3B) | ARCL3B (#614438) is caused by mutation in PYCR1 gene (179035) on chromosome 17q25. Inheritance is autosomal recessive. | ARCL3B is a rare autosomal recessive disorder characterized by a progeria-like appearance with distinctive facial features, sparse hair, ophthalmologic abnormalities and intrauterine growth retardation [168]. | |
| Cutis Laxa, autosomal recessive, Type IIIA (ARCL3A) | ARCL3A (#219150) is caused by mutation in the ALDH18A1 gene (*138250) on chromosome 10q24. Inheritance is autosomal recessive. | The protein encoded by ALDH18A1 catalyses the reduction of glutamate to delta1-pyrroline-5-carboxylate, a critical step in the de novo biosynthesis of proline, ornithine and arginine. | ARCL3A is characterized by cutis laxa (a progeria-like appearance) and ophthalmologic abnormalities [169]. In some case, additional features have been described, including delayed development, intellectual disability, seizures and problems with movement that can worsen over time. |
| Cutis Laxa, autosomal dominant, Type III (ADCL3) | ADCL3 (#616603) is caused by mutation in ALDH18A1 gene (*138250) on chromosome 10q24. Inheritance is autosomal dominant. | ADCL3 has a progeroid appearance characterized by thin skin with visible veins and wrinkles, ophthalmological abnormalities, clenched fingers, pre- and postnatal growth retardation and moderate intellectual disability. Patients also exhibit a combination of muscular hypotonia with brisk muscle reflexes [170]. | |
| Keratoconus-1 (KTCN1) | KTCN1 (#148300) is caused by heterozygous mutation in the Visual system homeobox gene 1 (VSX1, *605020) gene on 20p11 chromosome. Inheritance is autosomal dominant. | VSX1 encodes a homeoprotein that regulates the expression of the cone opsin genes early in development. Recent studies showed that the structural deformity of the cornea in KCTN patients may be due to reduced expression of collagens (COL1A1 and COL4A1) and LOX family oxidases, as well as on the concomitant increased expression of MMP9 [171]. | KTCN1 is the most common corneal dystrophy. It is a bilateral, often asymmetrical, non-inflammatory progressive corneal ectasia that causes visual morbidity. In affected individuals, the cornea becomes progressively thin and conical in shape, resulting in myopia, irregular astigmatism and corneal scarring. It typically appears in the teenage years and then it progresses until the third and fourth decades. No specific treatment exists except corneal transplantation when visual acuity can no longer be corrected by contact lenses [172]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arseni, L.; Lombardi, A.; Orioli, D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018, 19, 1407. https://doi.org/10.3390/ijms19051407
Arseni L, Lombardi A, Orioli D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. International Journal of Molecular Sciences. 2018; 19(5):1407. https://doi.org/10.3390/ijms19051407
Chicago/Turabian StyleArseni, Lavinia, Anita Lombardi, and Donata Orioli. 2018. "From Structure to Phenotype: Impact of Collagen Alterations on Human Health" International Journal of Molecular Sciences 19, no. 5: 1407. https://doi.org/10.3390/ijms19051407
APA StyleArseni, L., Lombardi, A., & Orioli, D. (2018). From Structure to Phenotype: Impact of Collagen Alterations on Human Health. International Journal of Molecular Sciences, 19(5), 1407. https://doi.org/10.3390/ijms19051407