cPKCγ-Modulated Sequential Reactivation of mTOR Inhibited Autophagic Flux in Neurons Exposed to Oxygen Glucose Deprivation/Reperfusion


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
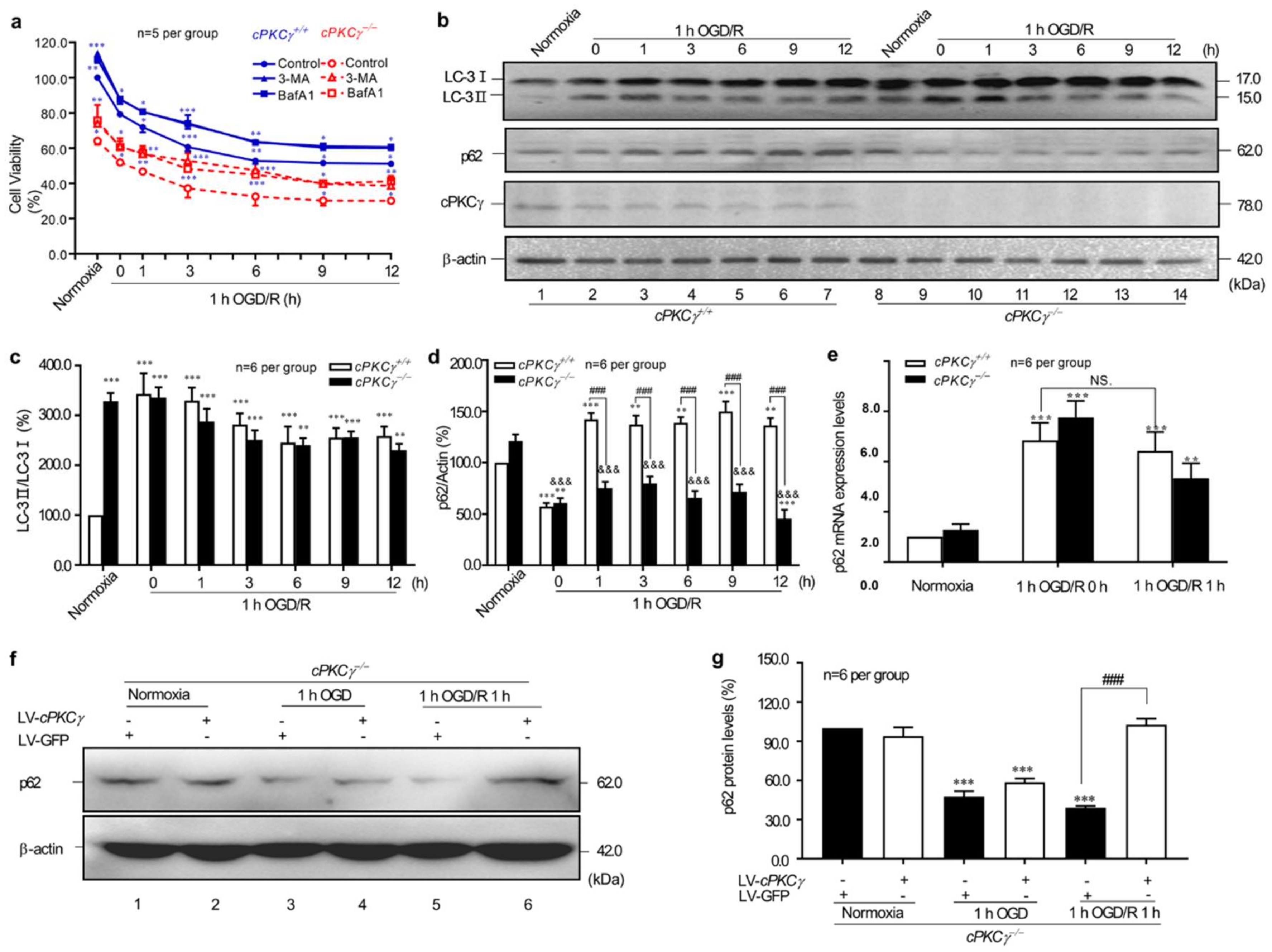
2.1. cPKCγ Gene Knockout Reduces the Survival of Primary Cortical Neurons after 1 h OGD/R 0–12 h Treatments through Excessive Autophagy
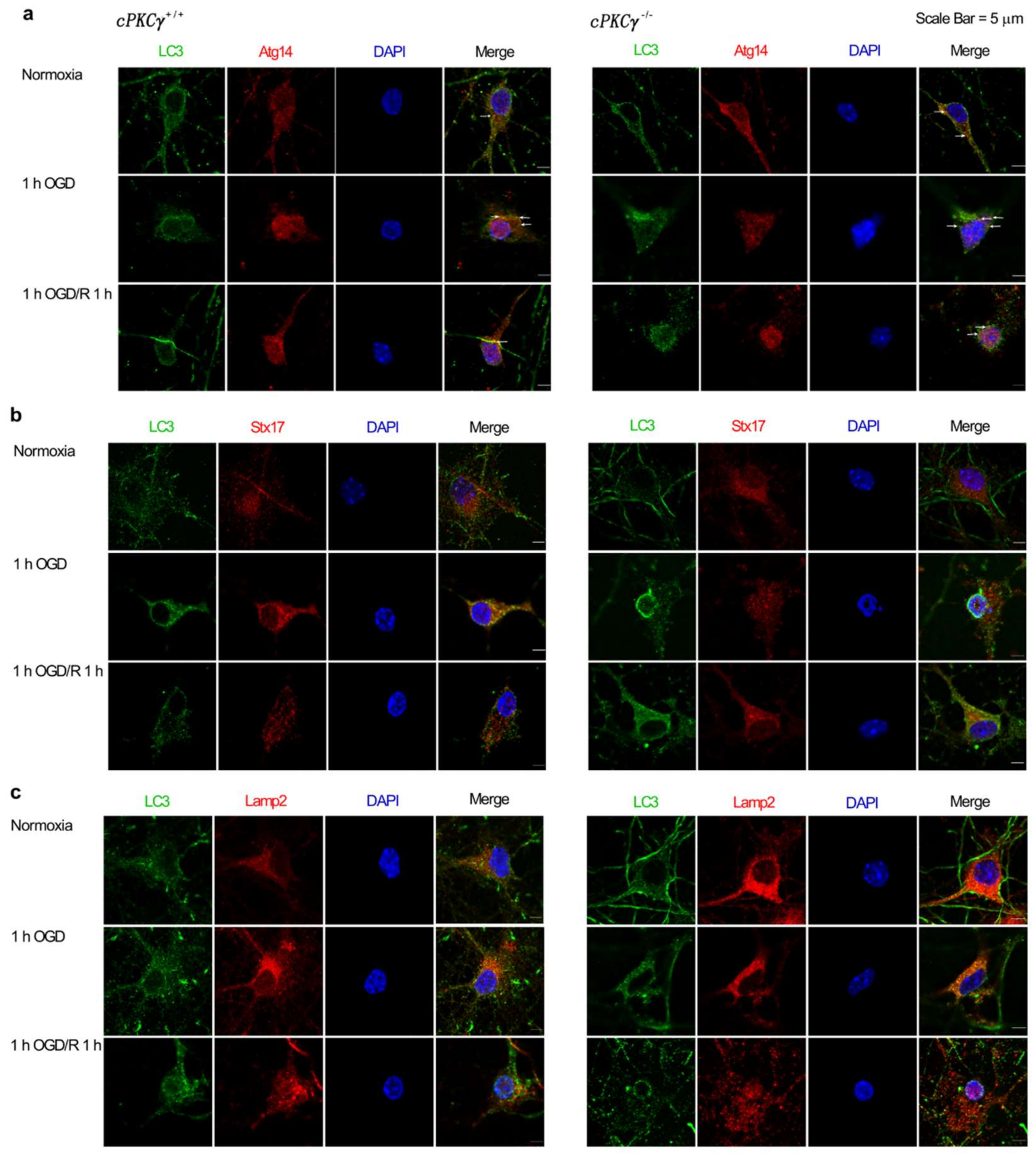
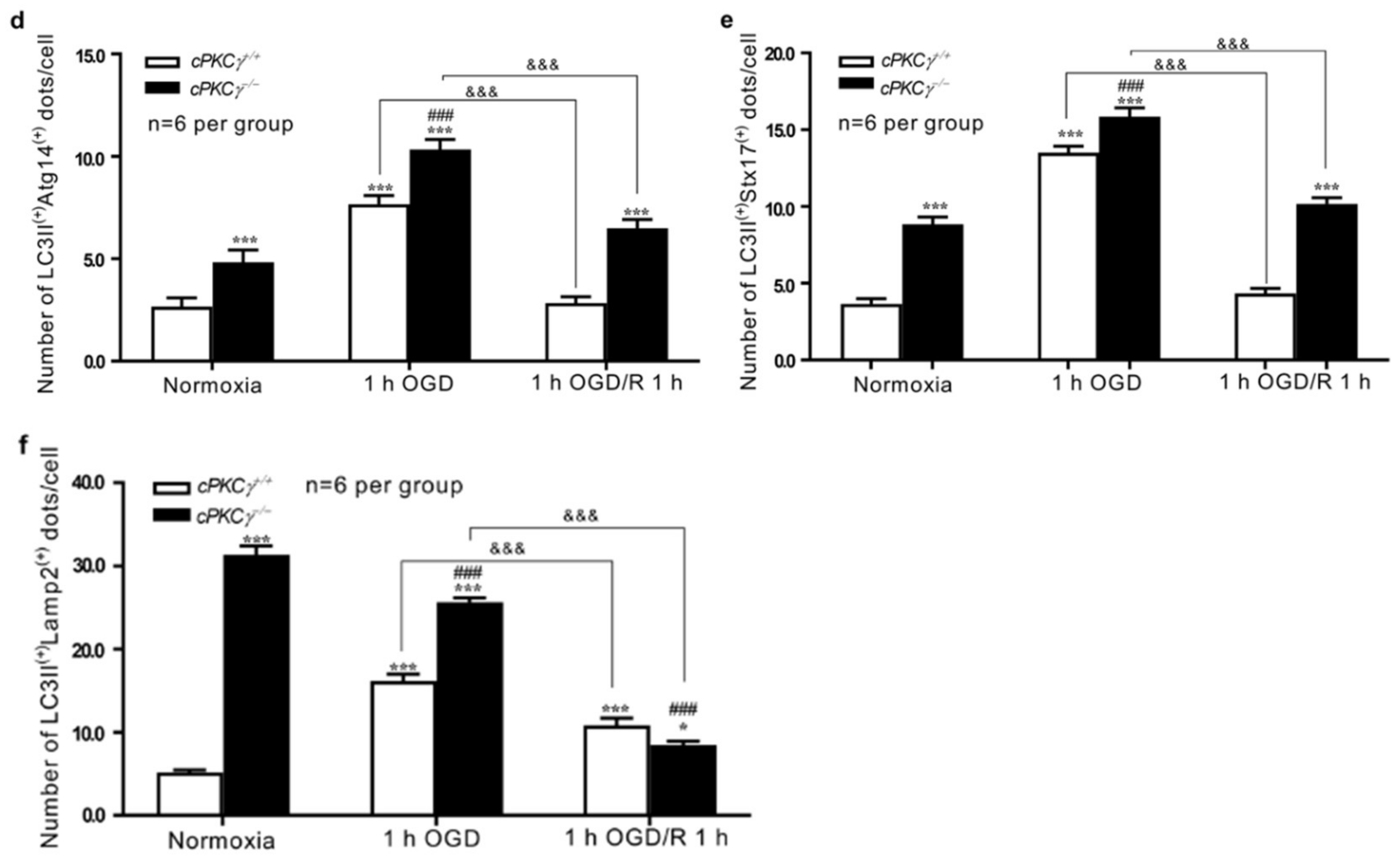
2.2. cPKCγ Partly Blocks Autophagic Flux in Neurons after 1 h OGD/R 0–12 h Treatments
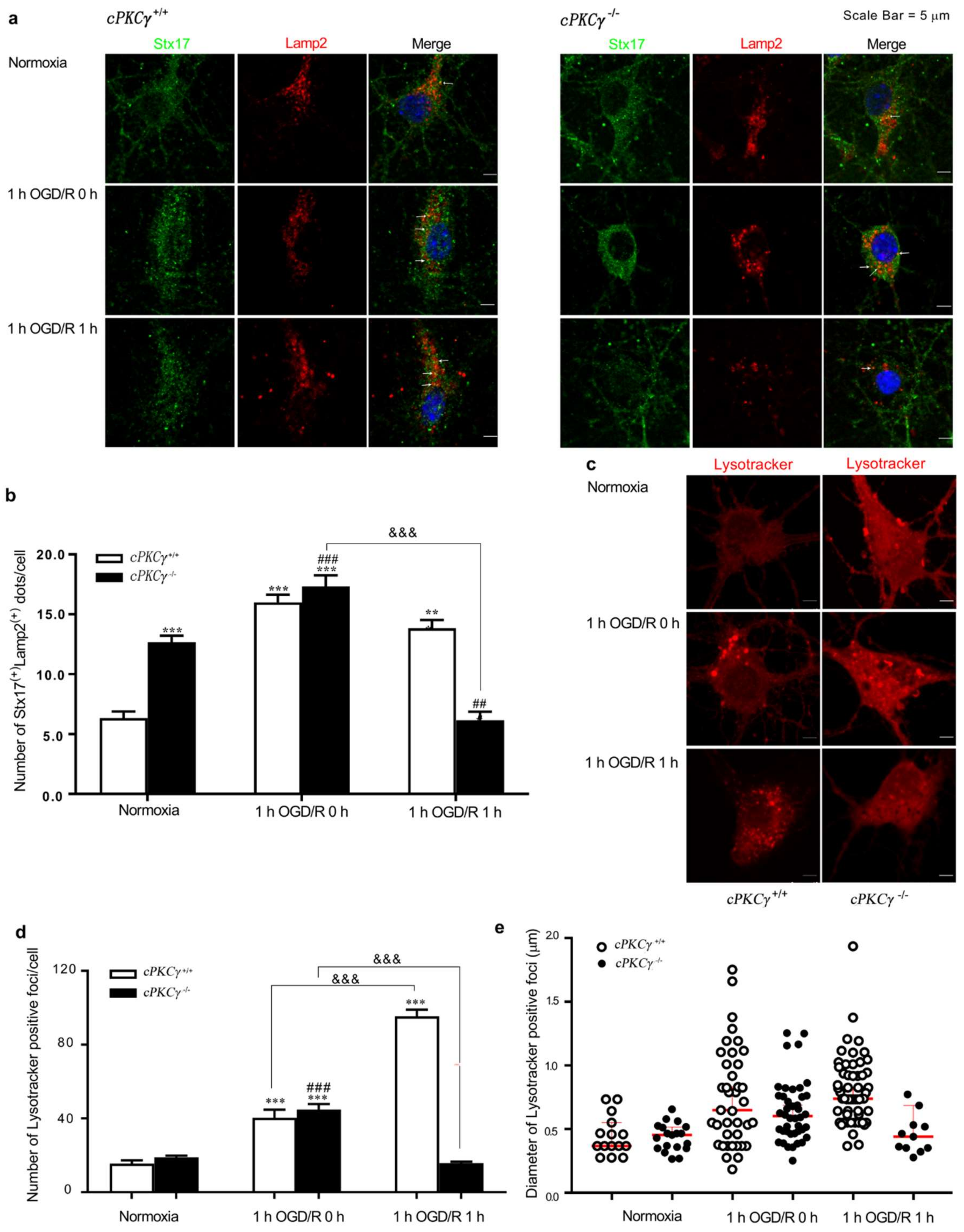
2.3. Effects of cPKCγ on Autolysosome Formation and Degradation in Neurons after 1 h OGD/R 0–1 h Treatments
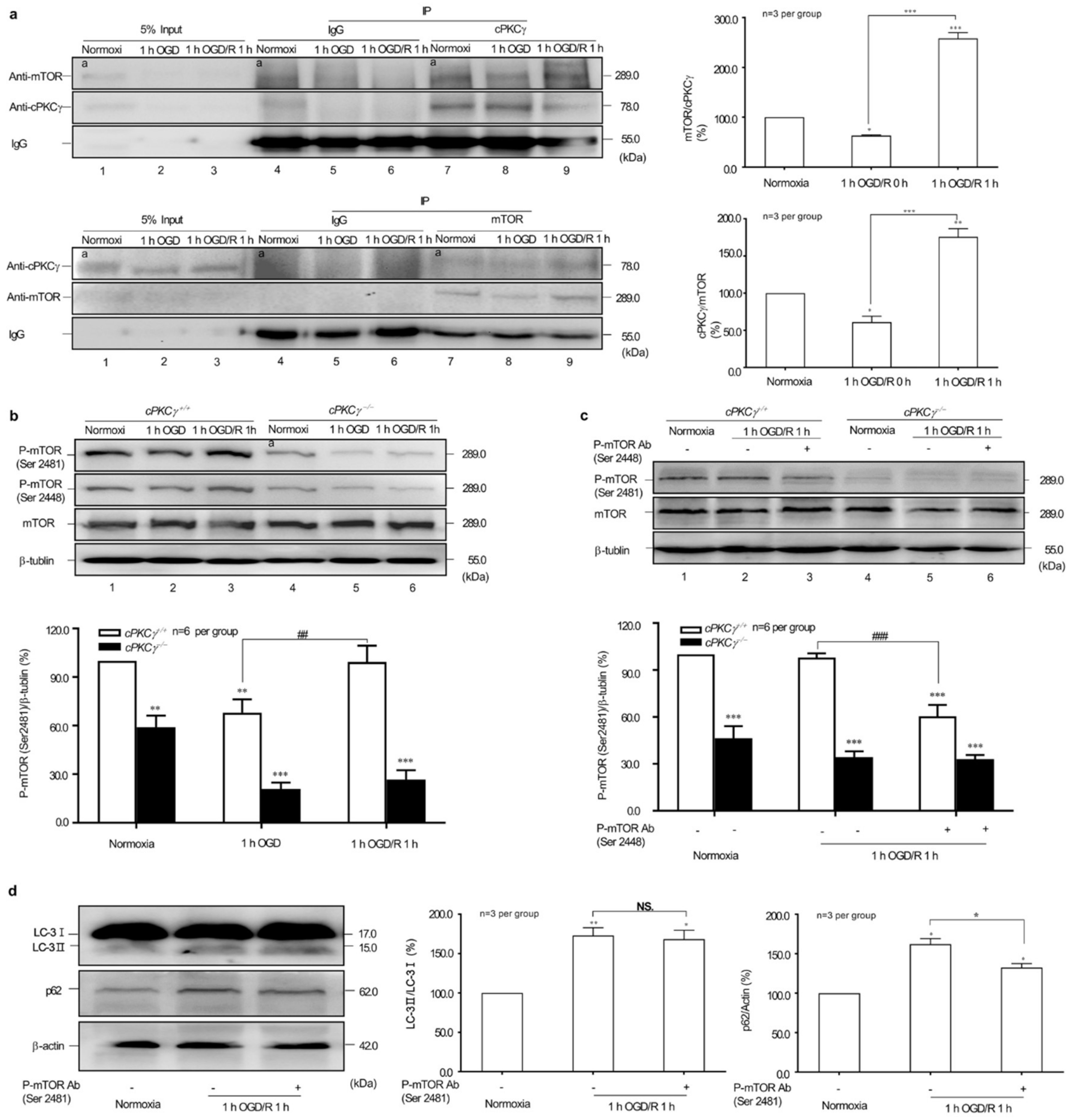
2.4. Effects of cPKCγ on mTOR Reactivation in 1 h OGD/R 1 h-Treated Neurons
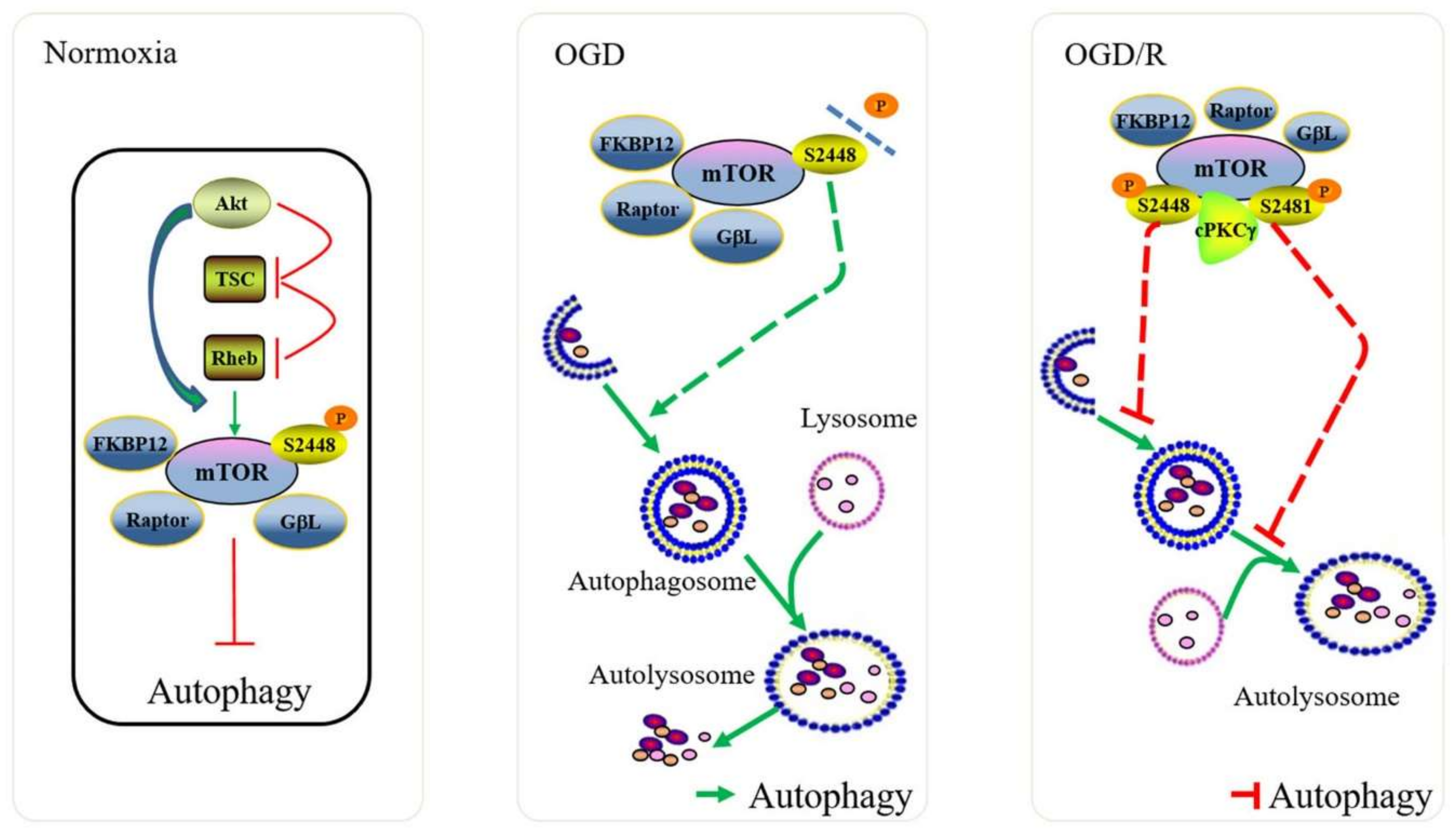
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Oxygen Glucose Deprivation/Reperfusion (OGD/R) of Primary Cortical Neurons
4.3. Antibodies
4.4. Cell Viability Test
4.5. Immunoprecipitation Assays
4.6. Immunofluorescence
4.7. RNA Extraction and Real-Time Quantitative PCR
4.8. Immunoblotting
4.9. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| cPKCγ | Conventional Protein Kinase Cγ |
| rtPA | recombinant tissue Plasminogen Activator |
| LC3 | Microtubule-associated Protein 1 Light Chain 3 |
| MCAO/R | Middle Cerebral Artery Occlusion/Reperfusion |
| mTOR | Mechanistic Target of Rapamycin |
| OGD/R | Oxygen Glucose Deprivation/Reperfusion |
| SQSTM1 | Sequestosome 1 |
| ALR | Autophagic Lysosome Reformation |
| BafA1 | Bafilomycin A1 |
| 3-MA | 6-Amino-3-methylpurine |
References
- Zhang, N.; Yin, Y.; Han, S.; Jiang, J.; Yang, W.; Bu, X.; Li, J. Hypoxic preconditioning induced neuroprotection against cerebral ischemic injuries and its cpkcgamma-mediated molecular mechanism. Neurochem. Int. 2011, 58, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, Y.; Han, S.; Liu, S.; Zhang, N.; Zhao, L.; Li, S.; Li, J. Cpkcgamma-modulated autophagy in neurons alleviates ischemic injury in brain of mice with ischemic stroke through akt-mtor pathway. Transl. Stroke Res. 2016, 7, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Subramani, S.; Malhotra, V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013, 14, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/sqstm1 binds directly to atg8/lc3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mtor. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. Atg14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Wartosch, L.; Gunesdogan, U.; Graham, S.C.; Luzio, J.P. Recruitment of vps33a to hops by vps16 is required for lysosome fusion with endosomes and autophagosomes. Traffic 2015, 16, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Li, W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the vps33a(d251e) mutation. Autophagy 2015, 11, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of mtorc1 by the rag gtpases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.G.; Puyal, J. Autophagic cell death exists. Autophagy 2012, 8, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, C.; Gao, K.; Zhao, H.; Zhou, Z.; Shen, Z.; Guo, Y.; Li, Z.; Yao, T.; Mei, X. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem. Biophys. Res. Commun. 2016, 477, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ho, A.; Lee, J.H. Autophagy and human neurodegenerative diseases—A fly’s perspective. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Tammineni, P.; Cai, Q. Defective retrograde transport impairs autophagic clearance in alzheimer disease neurons. Autophagy 2017, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mtor complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The mammalian ulk1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, L. Autophagic lysosome reformation. Exp. Cell Res. 2013, 319, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, M.; Hadley, G.; Xilouri, M.; Hoyte, L.C.; Nagel, S.; McMenamin, M.M.; Tsaknakis, G.; Watt, S.M.; Drakesmith, C.W.; Chen, R.; et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat. Med. 2013, 19, 351–357. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, R.; Han, S.; Zhang, N.; Dai, Q.; Liu, T.; Li, J. cPKCγ-Modulated Sequential Reactivation of mTOR Inhibited Autophagic Flux in Neurons Exposed to Oxygen Glucose Deprivation/Reperfusion. Int. J. Mol. Sci. 2018, 19, 1380. https://doi.org/10.3390/ijms19051380
Hua R, Han S, Zhang N, Dai Q, Liu T, Li J. cPKCγ-Modulated Sequential Reactivation of mTOR Inhibited Autophagic Flux in Neurons Exposed to Oxygen Glucose Deprivation/Reperfusion. International Journal of Molecular Sciences. 2018; 19(5):1380. https://doi.org/10.3390/ijms19051380
Chicago/Turabian StyleHua, Rongrong, Song Han, Nan Zhang, Qingqing Dai, Ting Liu, and Junfa Li. 2018. "cPKCγ-Modulated Sequential Reactivation of mTOR Inhibited Autophagic Flux in Neurons Exposed to Oxygen Glucose Deprivation/Reperfusion" International Journal of Molecular Sciences 19, no. 5: 1380. https://doi.org/10.3390/ijms19051380
APA StyleHua, R., Han, S., Zhang, N., Dai, Q., Liu, T., & Li, J. (2018). cPKCγ-Modulated Sequential Reactivation of mTOR Inhibited Autophagic Flux in Neurons Exposed to Oxygen Glucose Deprivation/Reperfusion. International Journal of Molecular Sciences, 19(5), 1380. https://doi.org/10.3390/ijms19051380