Mechanisms of Intrinsic Tumor Resistance to Immunotherapy



Abstract
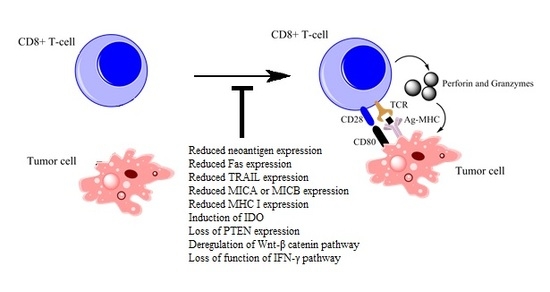
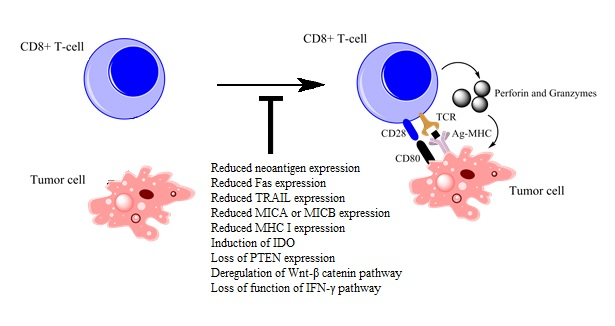
1. Introduction
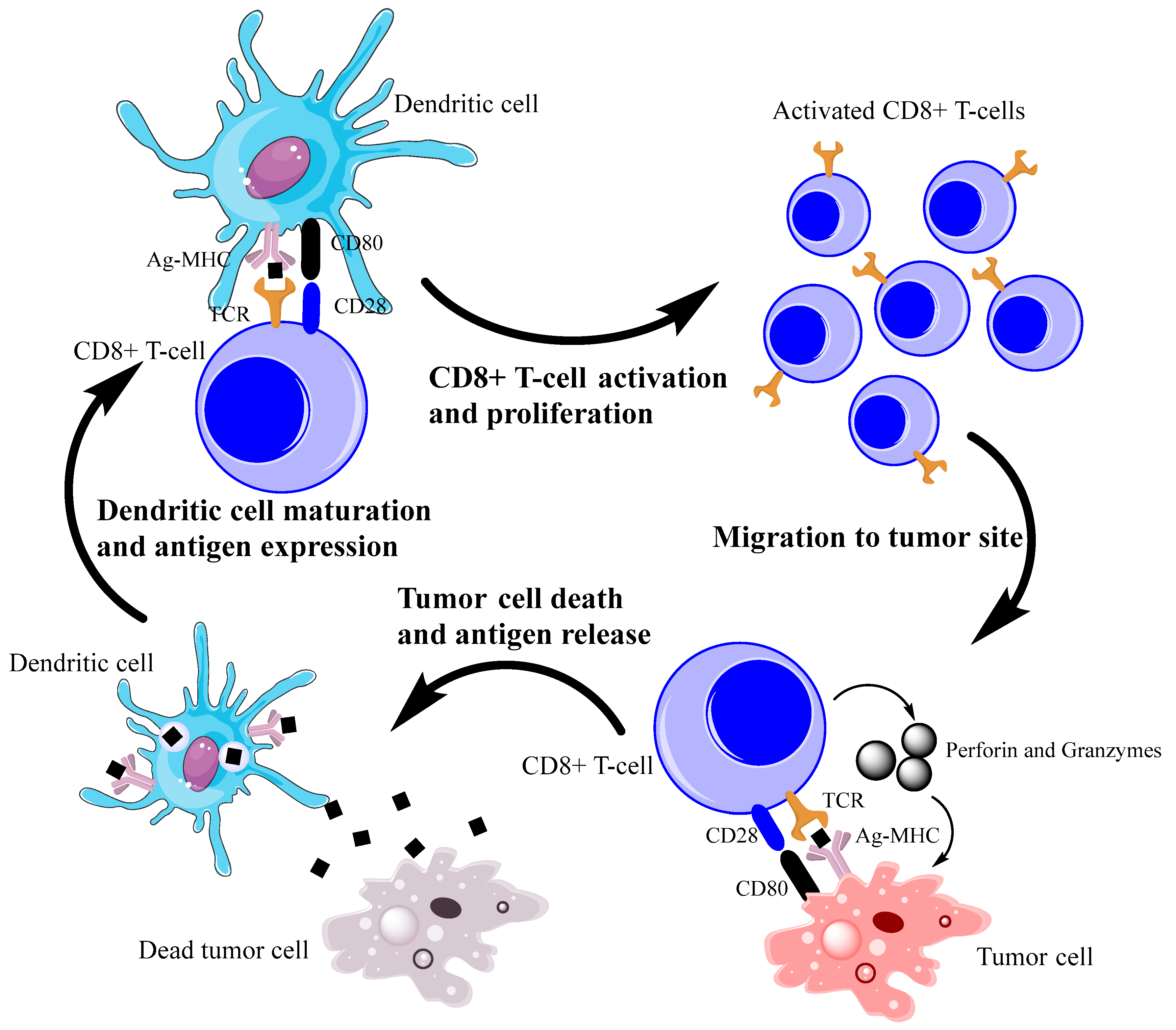
2. Antitumor Immune Response
3. Intrinsic Resistance Mechanisms
3.1. Reductions in Antigen Expression
3.2. Alterations in Cell Receptor Expression
3.3. Alterations in CD28 Expression
3.4. Alterations in Cellular Enzymes and Metabolic Pathways
3.4.1. Induction of IDO
3.4.2. Loss of PTEN Expression
3.4.3. Deregulated Expression of the Wnt–β-Catenin Pathway
3.5. Activation of the IFN-γ Pathway
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Holzinger, A.; Barden, M.; Abken, H. The growing world of CAR T cell trials: A systematic review. Cancer Immunol. Immunother. 2016, 65, 1433–1450. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; de Vries, I.J.M.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, Z.; Tang, L.; Xu, Y.C.; Xie, Z.M.; Gu, X.F.; Wang, H.X. Cytokine-induced killer cells in the treatment of patients with solid carcinomas: A systematic review and pooled analysis. Cytotherapy 2012, 14, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Smyth, M.J.; Teng, M.W. Acquired resistance to anti-PD1 therapy: Checkmate to checkpoint blockade? Genome Med. 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Oh, M.S.; Giles, F.J. Molecular Biomarkers of Primary and Acquired Resistance to T-Cell-Mediated Immunotherapy in Cancer: Landscape, Clinical Implications, and Future Directions. Oncologist 2018, 23, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar] [PubMed]
- Yukihirofunada, T.; Kikuchi, R.; Shinsuketakeno, Y.U.; Gabbert, H.E. Prognostic significance of CD8+ T cell and macrophage peritumoral infiltration in colorectal cancer. Oncol. Rep. 2003, 10, 309–313. [Google Scholar]
- Schumacher, K.; Haensch, W.; Röefzaad, C.; Schlag, P.M. Prognostic significance of activated CD8+ T cell infiltrations within esophageal carcinomas. Cancer Res. 2001, 61, 3932–3936. [Google Scholar] [PubMed]
- Kawai, O.; Ishii, G.; Kubota, K.; Murata, Y.; Naito, Y.; Mizuno, T.; Aokage, K.; Saijo, N.; Nishiwaki, Y.; Gemma, A. Predominant infiltration of macrophages and CD8+ T cells in cancer nests is a significant predictor of survival in stage IV nonsmall cell lung cancer. Cancer 2008, 113, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Coukos, G. Deciphering and reversing tumor immune suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Page, D.B.; Ku, G.Y.; Li, Y.; Mu, Z.; Ariyan, C.; Gallardo, H.F.; Roman, R.-A.; Heine, A.I.; Terzulli, S.L. Correlation of clinical and immunological data in a metastatic melanoma patient with heterogeneous tumor responses to ipilimumab therapy. Cancer Immunity Arch. 2010, 10, 1. [Google Scholar]
- Jäger, E.; Ringhoffer, M.; Altmannsberger, M.; Arand, M.; Karbach, J.; Jäger, D.; Oesch, F.; Knuth, A. Immunoselection in vivo: Independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. Int. J. Cancer 1997, 71, 142–147. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Blades, R.A.; Keating, P.J.; McWilliam, L.J.; George, N.J.; Stern, P.L. Loss of HLA class I expression in prostate cancer: Implications for immunotherapy. Urology 1995, 46, 681–687. [Google Scholar] [CrossRef]
- Watson, N.F.; Ramage, J.M.; Madjd, Z.; Spendlove, I.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int. J. Cancer 2006, 118, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742. [Google Scholar] [PubMed]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Franco, A.; Myers, K.; Gray, C.; Nguyen, T.; Hersey, P. Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma. Cancer Res. 1999, 59, 2747–2753. [Google Scholar] [PubMed]
- Pardoll, D. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 2003, 21, 807–839. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, W.S.; Zhu, W.F.; Lin, J.; Zhou, Z.F.; Huang, C.Z.; Chen, G.; Shi, Y.; Guo, Z.Q.; Ye, Y.B. Tumor MICA status predicts the efficacy of immunotherapy with cytokine-induced killer cells for patients with gastric cancer. Immunol. Res. 2016, 64, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Okita, R.; Yukawa, T.; Nojima, Y.; Maeda, A.; Saisho, S.; Shimizu, K.; Nakata, M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2016, 65, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Watson, N.F.; Spendlove, I.; Madjd, Z.; McGilvray, R.; Green, A.R.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Expression of the stress-related MHC class I chain-related protein MICA is an indicator of good prognosis in colorectal cancer patients. Int. J. Cancer 2006, 118, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Rammensee, H.-G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.Y.; Wang, P.Y.; Zhang, C.; Zhang, Y.L.; Fu, Y.; Zhang, C.; Li, Q.X.; Zhou, J.N.; Shan, B.E.; He, D.W. All-trans retinoic acid enhances cytotoxicity of CIK cells against human lung adenocarcinoma by upregulating MICA and IL-2 secretion. Sci. Rep. 2017, 7, 16481. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Groh, V.; Spies, T.; Kimura, R.; Miyagi, T.; Mochizuki, K.; Sasaki, Y. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int. J. Cancer 2003, 104, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Nwangwu, C.A.; Weiher, H.; Schmidt Wolf, I.G. Increase of CIK cell efficacy by upregulating cell surface MICA and inhibition of NKG2D ligand shedding in multiple myeloma. Hematol. Oncol. 2017, 35, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-X.; Zhang, W.G.; Chen, Y.X.; Zhao, W.H.; Tian, W.; Yang, Y.; Liu, S.H. Indoleamine 2, 3-dioxygenase expression in cells of human acute monocyte leukemia (M (5)) and acute lymphocyte leukemia and therapeutic effect of its inhibitor 1-methyl tryptophan. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2007, 15, 478–482. [Google Scholar] [PubMed]
- Curti, A.; Aluigi, M.; Pandolfi, S.; Ferri, E.; Isidori, A.; Salvestrini, V.; Durelli, I.; Horenstein, A.; Fiore, F.; Massaia, M. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia 2007, 21, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Schroecksnadel, K.; Winkler, C.; Fuith, L.C.; Fuchs, D. Tryptophan degradation in patients with gynecological cancer correlates with immune activation. Cancer Lett. 2005, 223, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Monjazeb, A.M.; Kent, M.S.; Grossenbacher, S.K.; Mall, C.; Zamora, A.E.; Mirsoian, A.; Chen, M.; Kol, A.; Shiao, S.L.; Reddy, A. Blocking indolamine-2,3-dioxygenase [sic] rebound immune suppression boosts antitumor effects of radio-immunotherapy in murine models and spontaneous canine malignancies. Clin. Cancer Res. 2016, 22, 4328–4340. [Google Scholar] [CrossRef] [PubMed]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Sautes-Fridman, C.; Tartour, E.; Kennedy, E.P.; Platten, M.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2014, 3, e957994. [Google Scholar] [CrossRef] [PubMed]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The role of indoleamine-2,3-dioxygenase (IDO) in cancer development, diagnostics, and therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Waldron, J.S.; Yang, I.; Han, S.; Tihan, T.; Sughrue, M.E.; Mills, S.A.; Pieper, R.O.; Parsa, A.T. Implications for immunotherapy of tumor-mediated T-cell apoptosis associated with loss of the tumor suppressor PTEN in glioblastoma. J. Clin. Neurosci. 2010, 17, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic [bgr]-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Irving, S.G.; Zipfel, P.F.; Balke, J.; McBride, O.W.; Morton, C.C.; Burd, P.R.; Siebenlist, U.; Kelly, K. Two inflammatory mediator cytokine genes are closely linked and variably amplified on chromosome 17q. Nucleic Acids Res. 1990, 18, 3261. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G. Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and-extrinsic factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.; Mok, T.S.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Bullani, R.; Wehrli, P.; Viard-Leveugle, I.; Rimoldi, D.; Cerottini, J.-C.; Saurat, J.-H.; Tschopp, J.; French, L. Frequent downregulation of Fas (CD95) expression and function in melanoma. Melanoma Res. 2002, 12, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Zhang, X.D.; Hersey, P. Relative resistance of fresh isolates of melanoma to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Clin. Cancer Res. 2001, 7, 966s–973s. [Google Scholar] [PubMed]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.K.; Robinson, B.W.; Lake, R.A. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003, 63, 4490–4496. [Google Scholar] [PubMed]
- King, G.D.; Muhammad, A.G.; Larocque, D.; Kelson, K.R.; Xiong, W.; Liu, C.; Sanderson, N.S.; Kroeger, K.M.; Castro, M.G.; Lowenstein, P.R. Combined Flt3L/TK gene therapy induces immunological surveillance which mediates an immune response against a surrogate brain tumor neoantigen. Mol. Therapy 2011, 19, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G.; Galluzzi, L. First Oncolytic Virus Approved for Melanoma Immunotherapy. Oncoimmunology 2016, 1, e1115641. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.; Seetharam, R.; Tesfa, L.; Augustine, T.A.; Klampfer, L.; Coffey, M.C.; Mariadason, J.M.; Goel, S. Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget 2014, 5, 2807–2819. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenet. 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.; Italiano, A.; Cleton-Jansen, A.-M.; Bovée, J.V. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gu, S.; Yin, M.; Shi, M.; Xin, C.; Zhu, J.; Wang, J.; Huang, S.; Xie, C.; Ma, J. Analysis of infantile fibrosarcoma reveals extensive T-cell responses within tumors: Implications for immunotherapy. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, T.S.; Akiyama, R.; Huang, R.R.; Shintaku, I.P.; Wang, X.; Tumeh, P.C.; Singh, A.; Chmielowski, B.; Denny, C.; Federman, N. Infiltration of CD8 T cells and expression of PD-1 and PD-L1 in synovial sarcoma. Cancer Immunol. Res. 2017, 5, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Sundara, Y.T.; Kostine, M.; Cleven, A.H.; Bovée, J.V.; Schilham, M.W.; Cleton-Jansen, A.-M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: A rationale for T-cell-based immunotherapy. Cancer Immunol. Immunother. 2017, 66, 119–128. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Mechanism | Examples of Associated Cancers | Citation |
|---|---|---|
| Reduced neoantigen generation | colorectal, kidney clear cell | Rooney et al. (2015) [62] |
| Reduced Fas expression | melanoma | Bullani et al. (2002) [63] |
| Reduced TRAIL expression | melanoma | Nguyen et al. (2001) [64] |
| Reduced MICA or MICB expression | colorectal, gastric | Salih et al. (2002) [35] |
| Reduced MHC I expression | melanoma, lung, breast, renal, prostate, bladder | Campoli et al. (2008) [65] |
| Induction of IDO | acute myeloid leukemia, colorectal, endometrial, small cell lung, melanoma, ovarian | Moon et al. (2015) [66] |
| Loss of PTEN expression | melanoma | Peng et al. (2016) [51] |
| Deregulation of Wnt–β-catenin pathway | melanoma | Spranger et al. (2015) [54] |
| Loss of function of IFN-γ pathway | melanoma, colorectal | Shin et al. (2017) [59] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rieth, J.; Subramanian, S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. Int. J. Mol. Sci. 2018, 19, 1340. https://doi.org/10.3390/ijms19051340
Rieth J, Subramanian S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. International Journal of Molecular Sciences. 2018; 19(5):1340. https://doi.org/10.3390/ijms19051340
Chicago/Turabian StyleRieth, John, and Subbaya Subramanian. 2018. "Mechanisms of Intrinsic Tumor Resistance to Immunotherapy" International Journal of Molecular Sciences 19, no. 5: 1340. https://doi.org/10.3390/ijms19051340
APA StyleRieth, J., & Subramanian, S. (2018). Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. International Journal of Molecular Sciences, 19(5), 1340. https://doi.org/10.3390/ijms19051340