Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation

 and
and 
Abstract
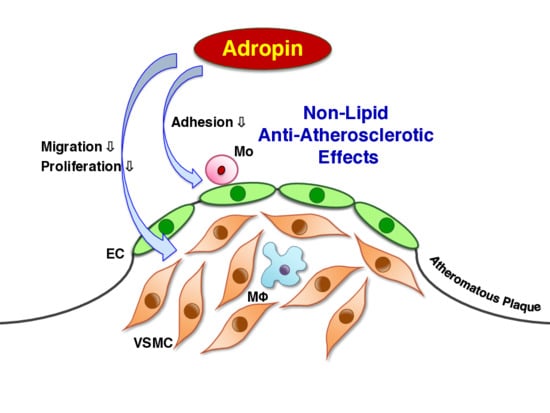
:
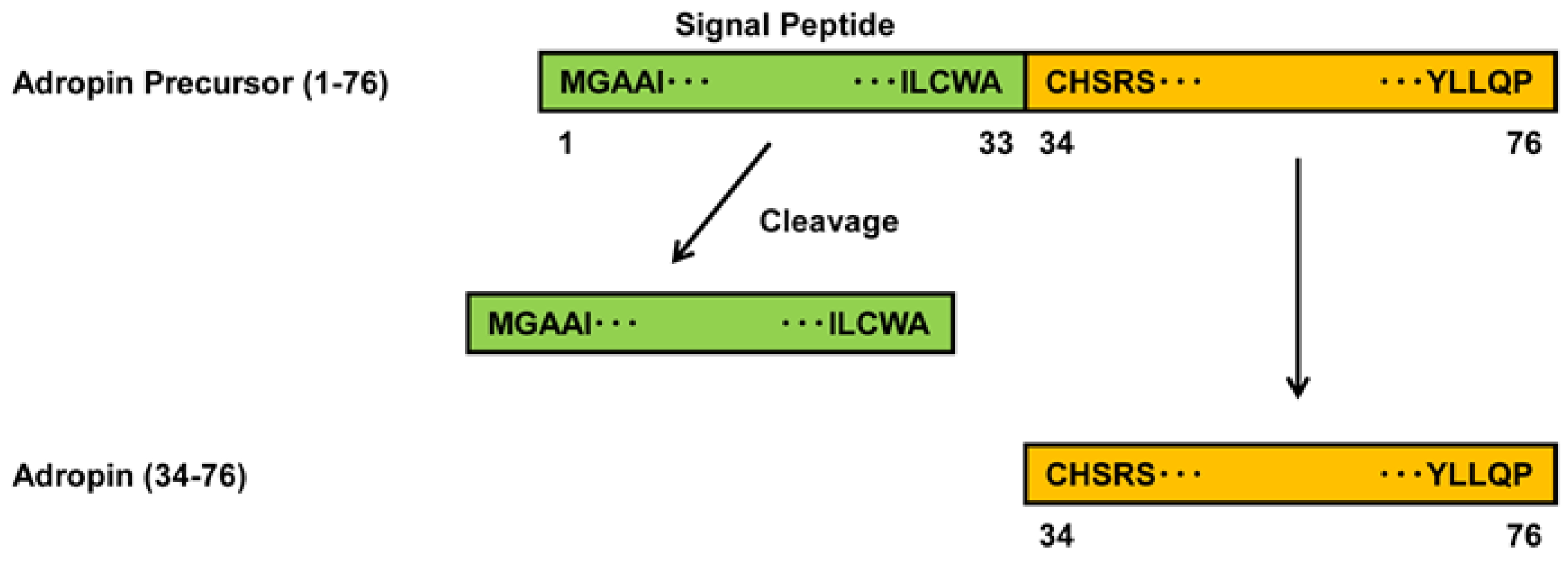
1. Introduction
2. Results
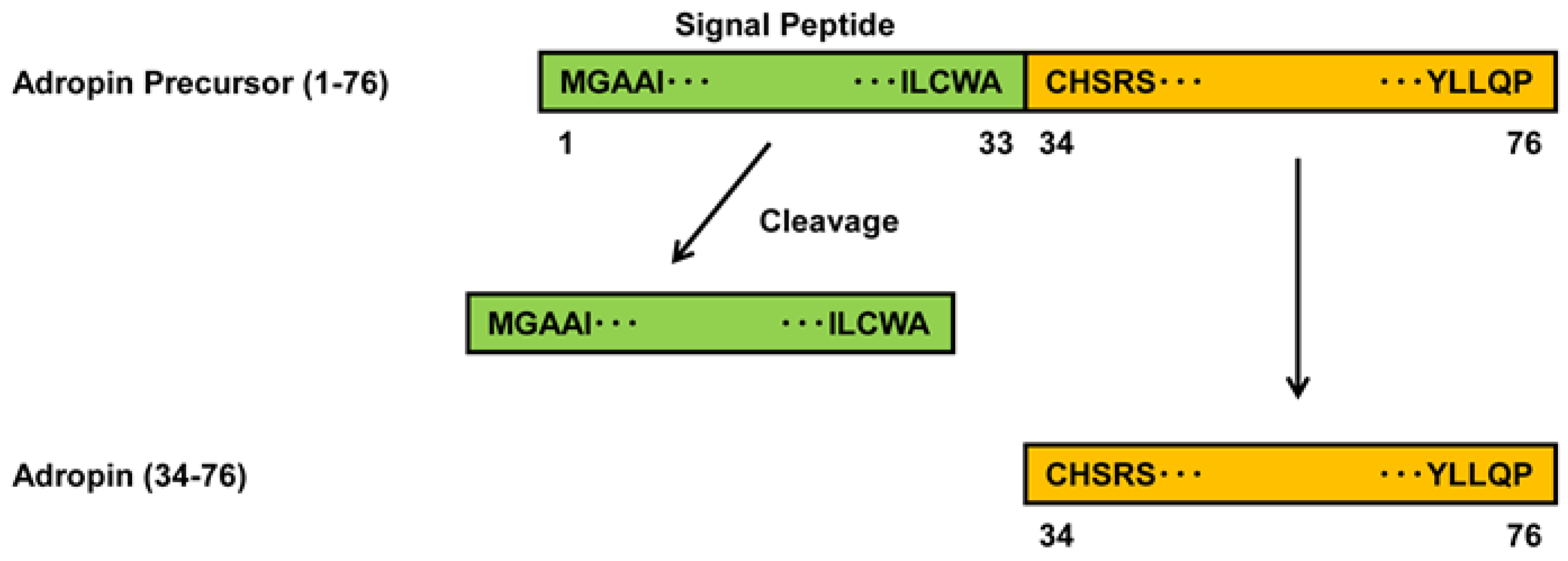
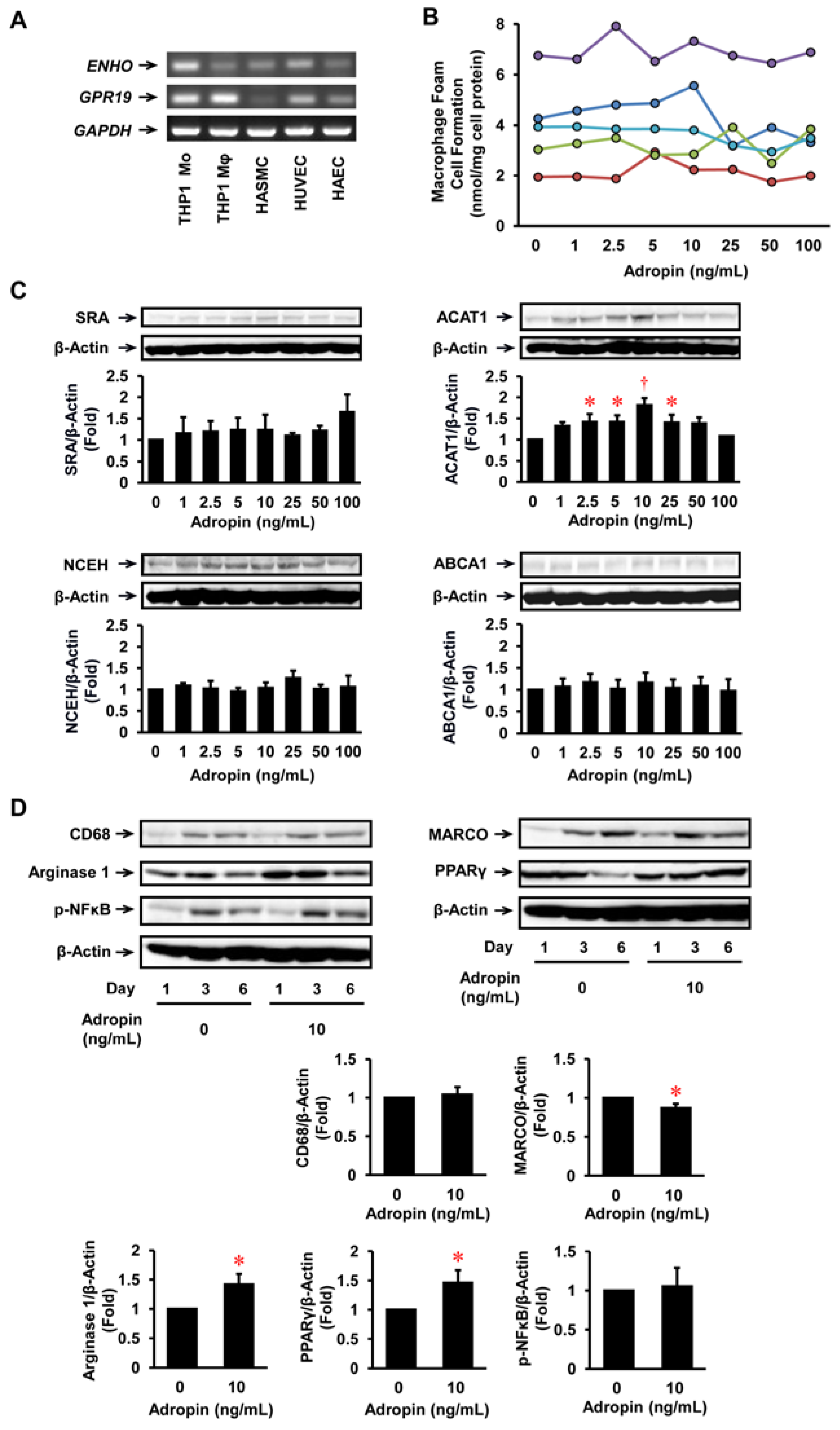
2.1. Expression of Adropin in Human Vascular Cells
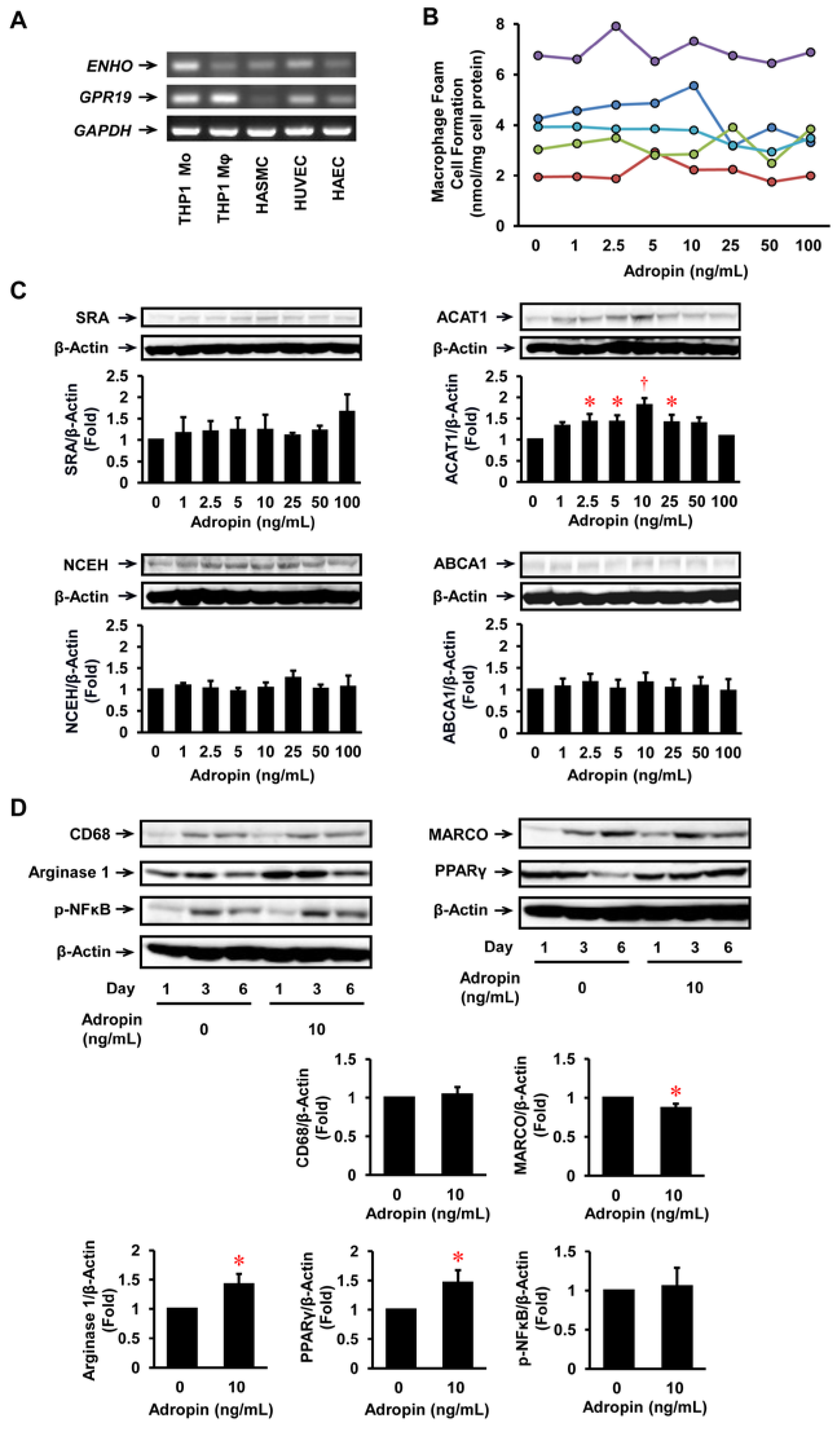
2.2. Effects of Adropin on Foam Cell Formation in Human Monocyte-Derived Macrophages
2.3. Effects of Adropin on Inflammatory Phenotypes in Human Monocyte-Derived Macrophages
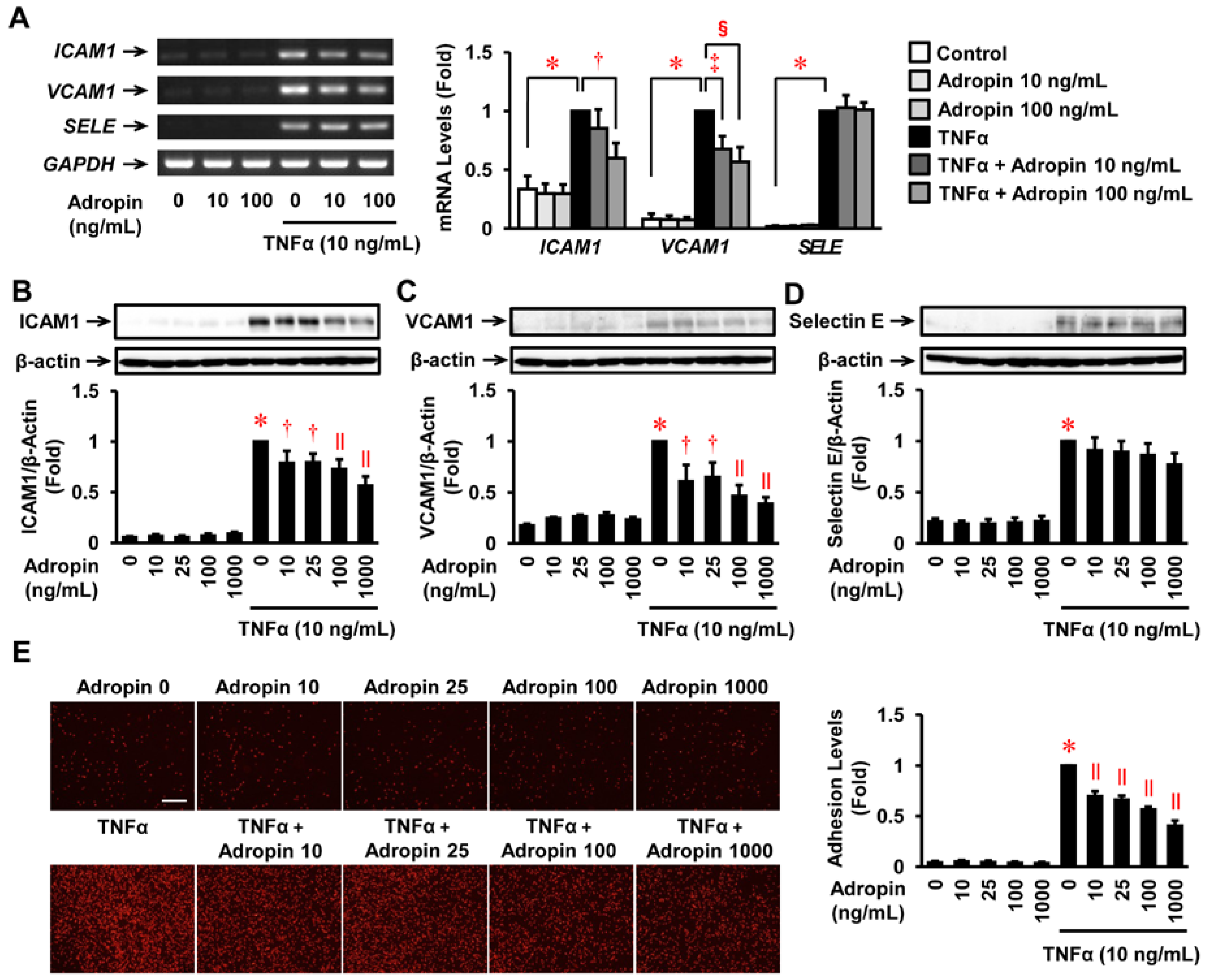
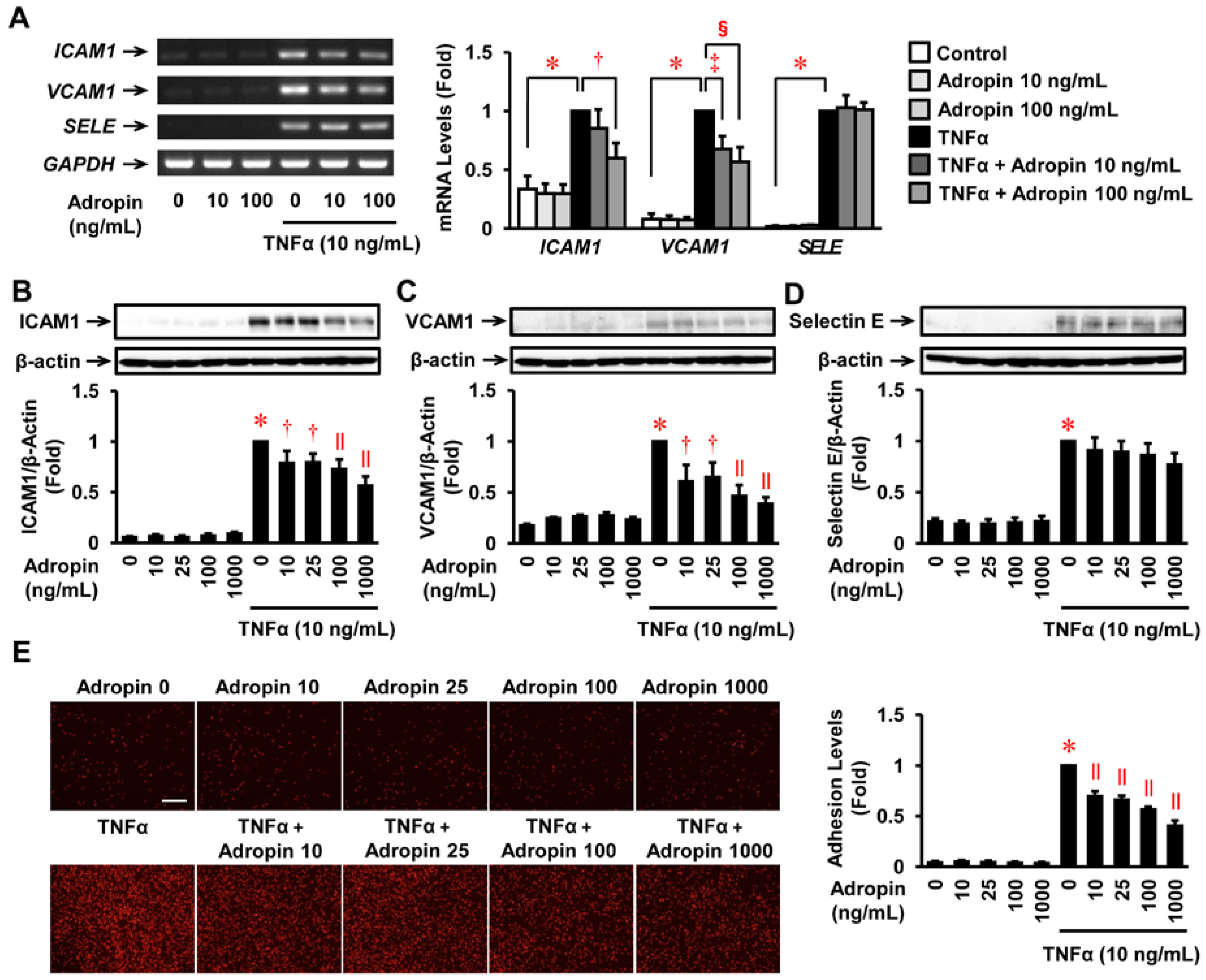
2.4. Effects of Adropin on the Inflammatory Response in Human ECs
2.5. Effects of Adropin on Human Monocyte Adhesion to Human ECs
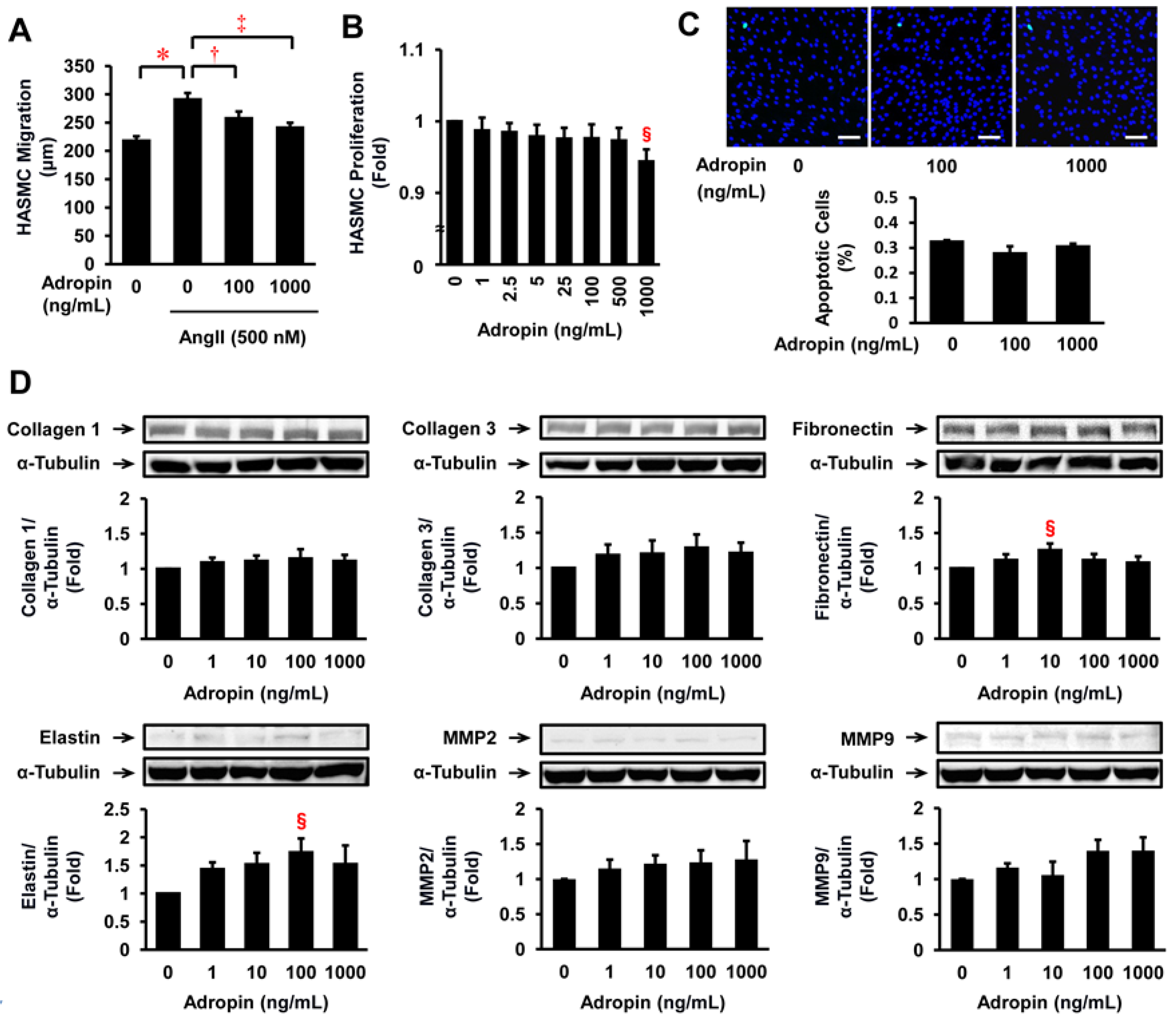
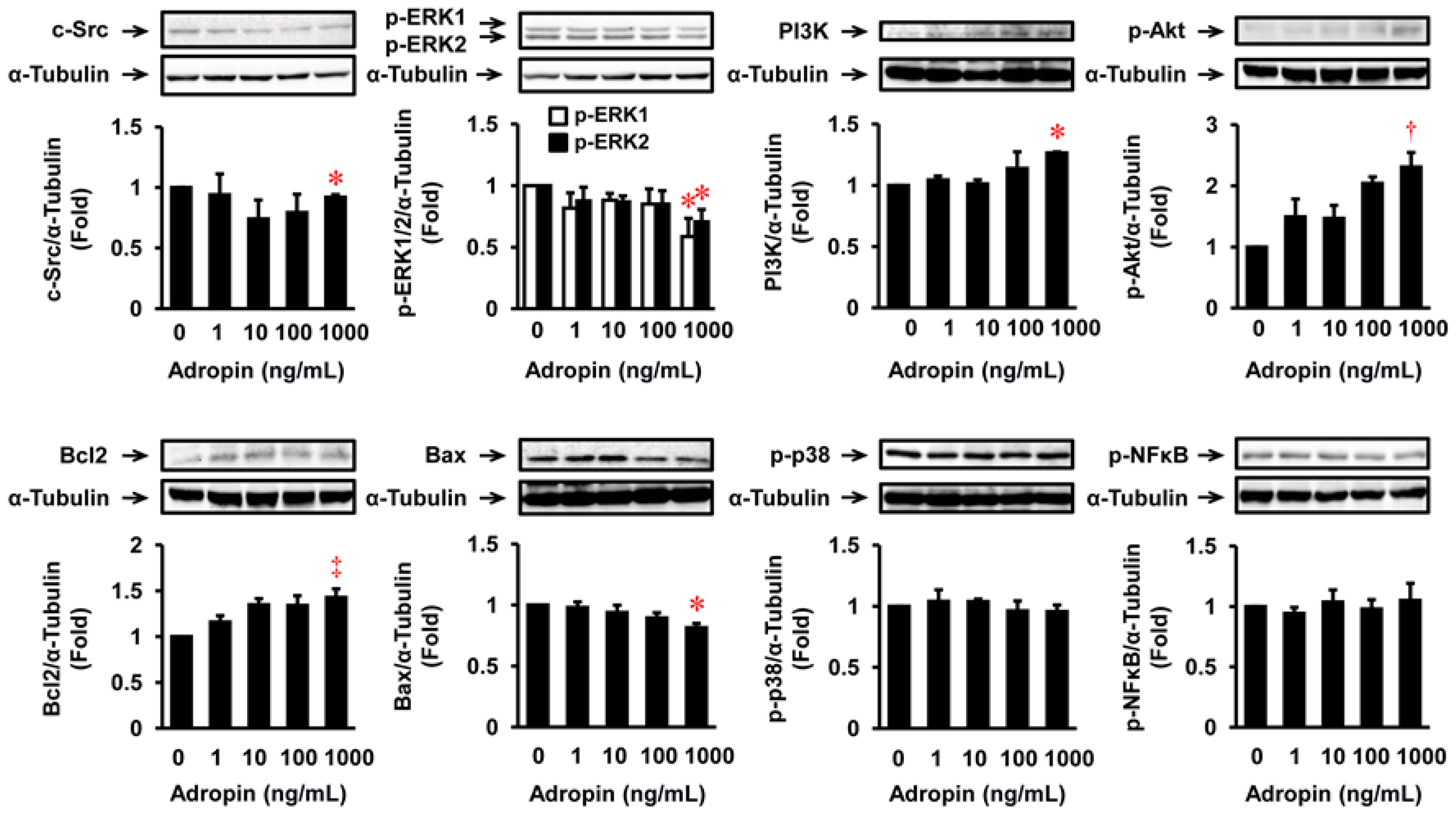
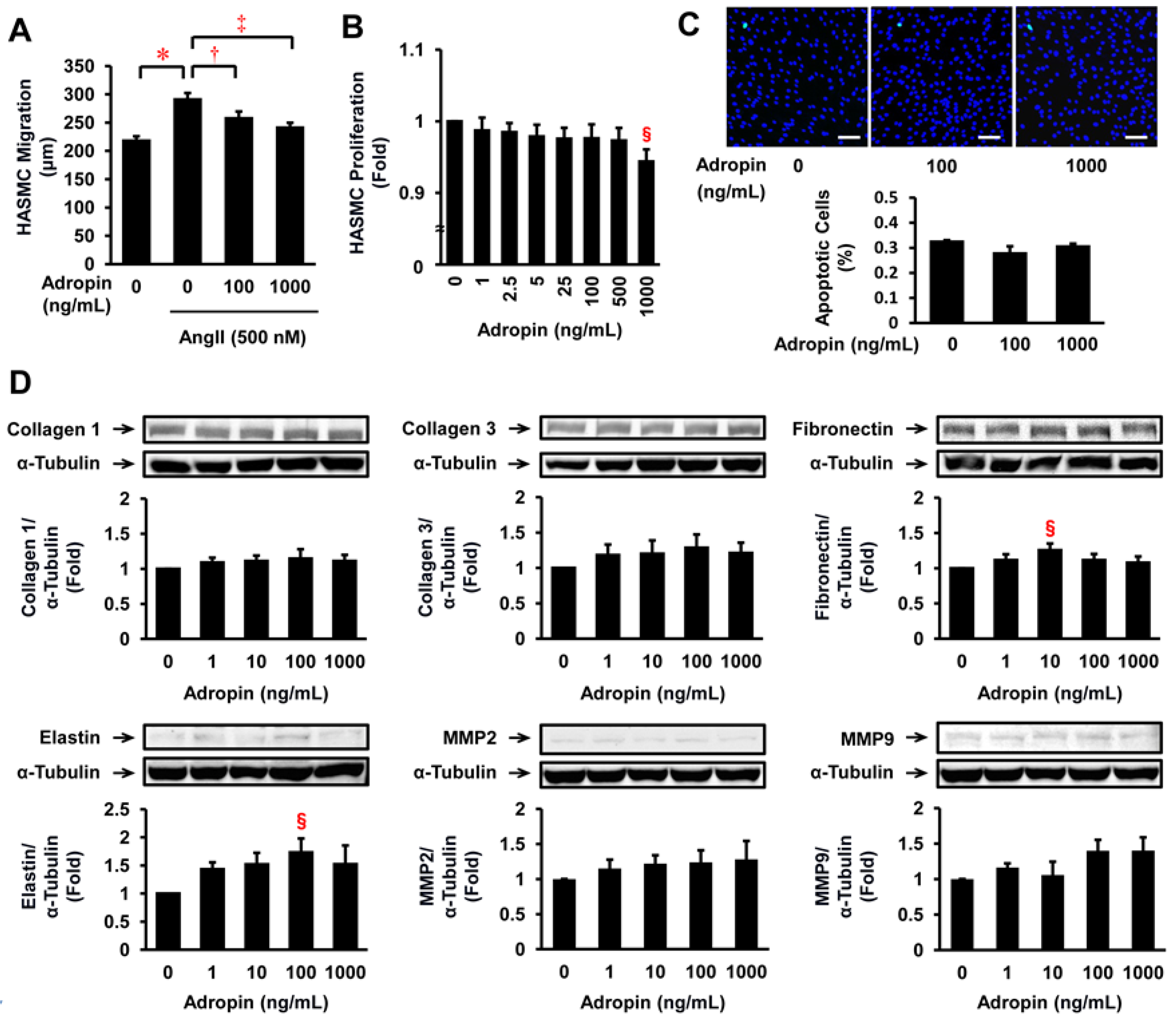
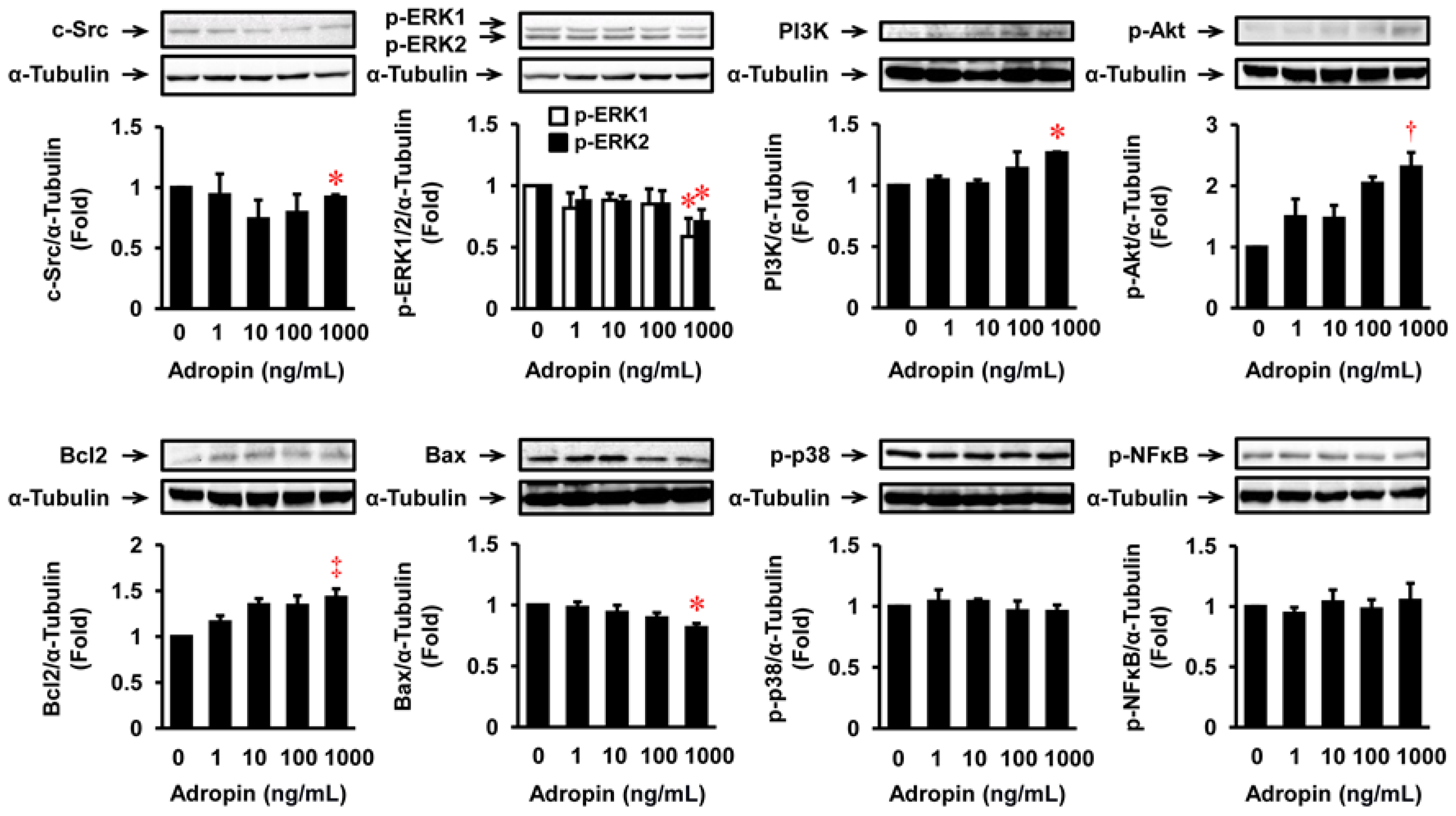
2.6. Effects of Adropin on Migration, Proliferation, Apoptosis, ECM Expression, and Relevant Signal Transduction in HASMCs
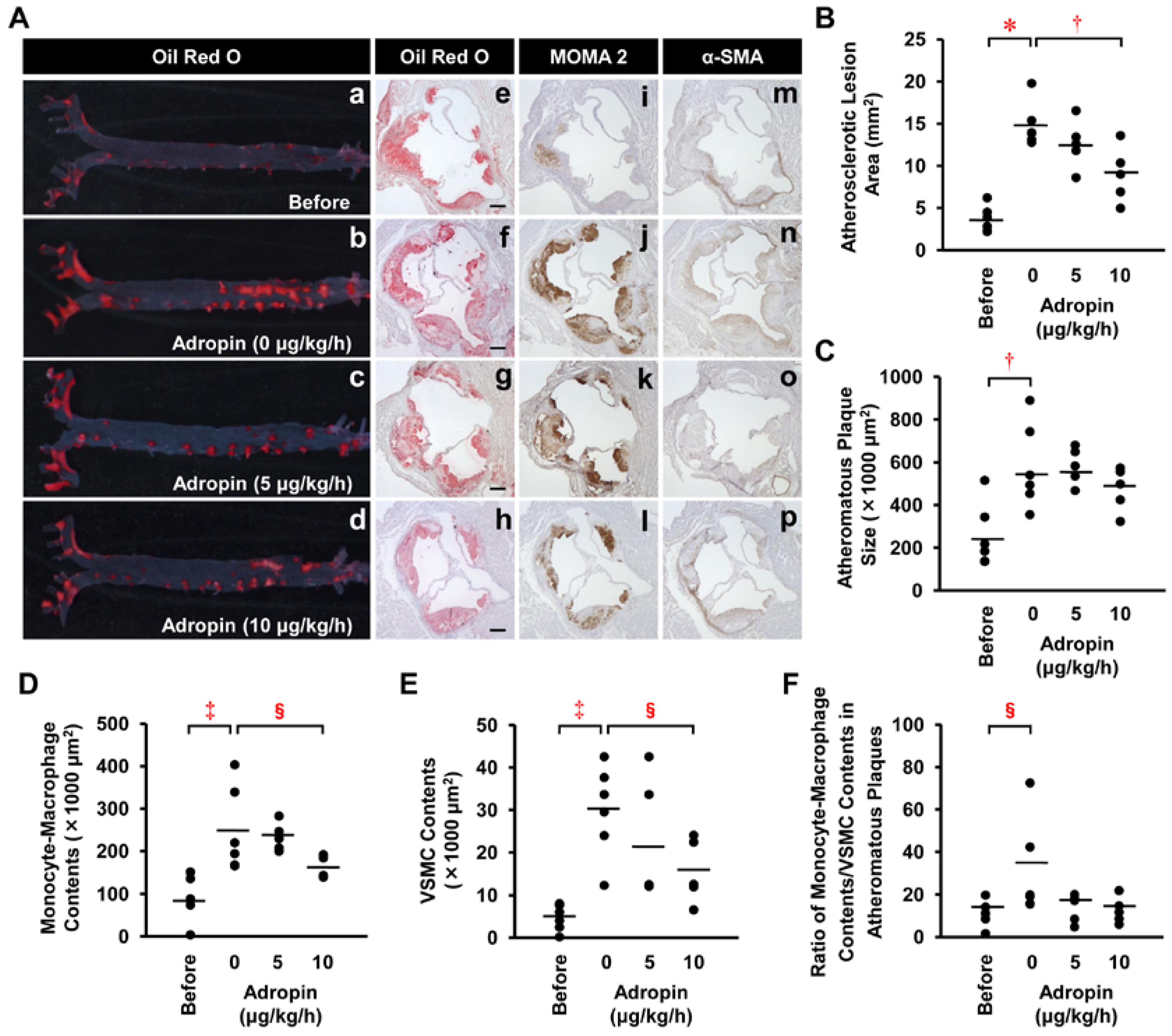
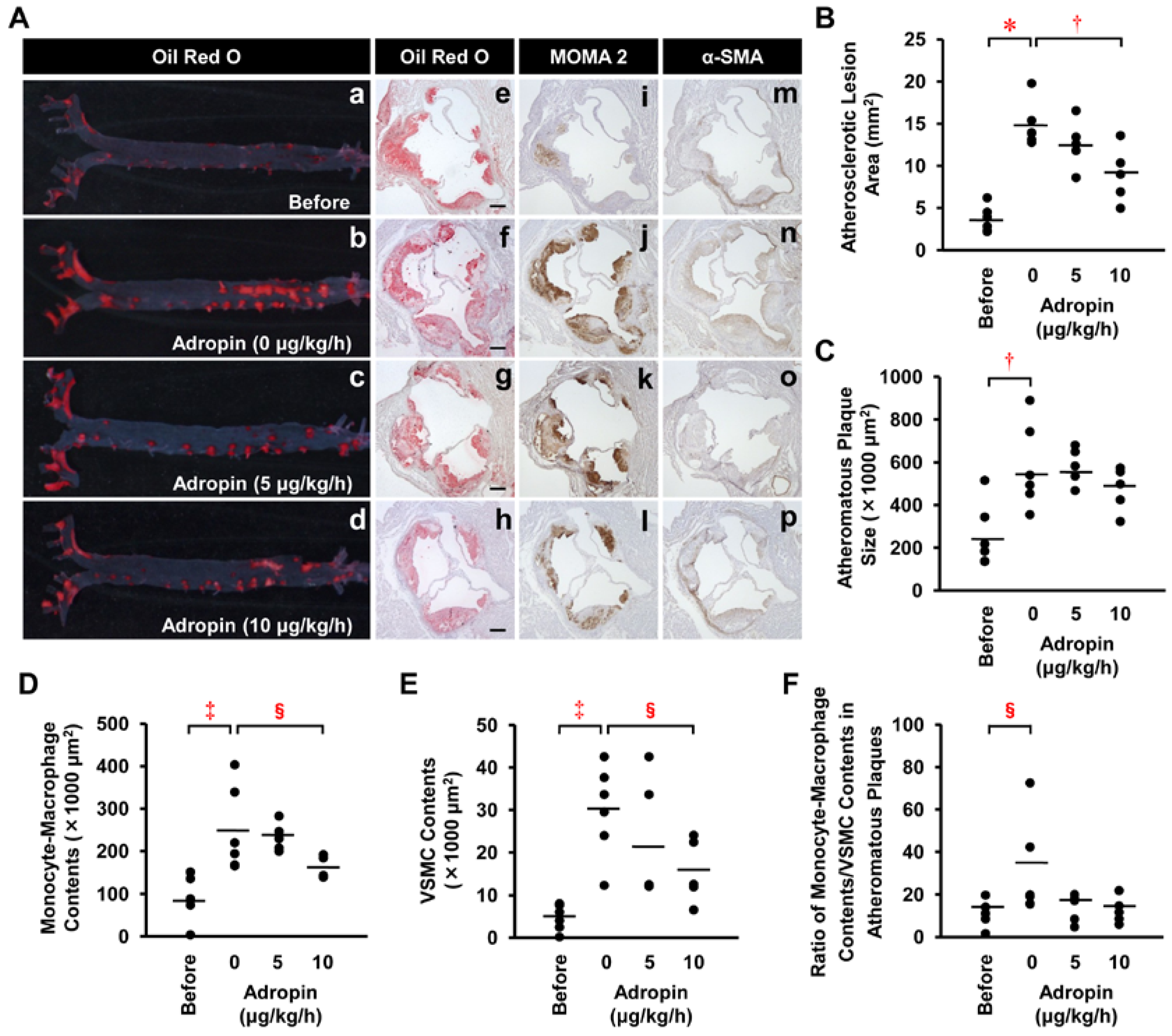
2.7. Effects of Adropin on Atherosclerotic Lesion Development in Apoe−/− Mice
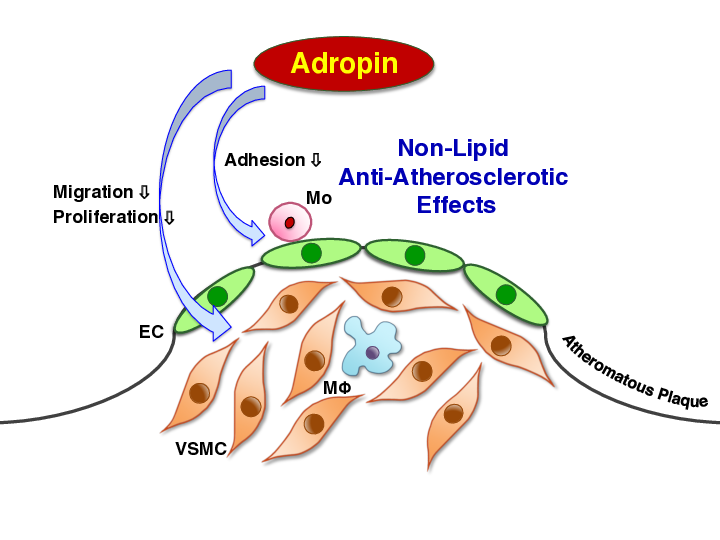
3. Discussion
4. Materials and Methods
4.1. Foam Cell Formation Assay
4.2. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.3. Monocyte Adhesion Assay
4.4. Migration Assay
4.5. Proliferation (Viability) Assay
4.6. Apoptosis Assay
4.7. Western Blotting
4.8. Animal Experiments
4.9. Animal Measurements
4.10. Assessment of Atherosclerotic Lesions
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter A1 |
| ACAT1 | acyl-coenzyme A: cholesterol acyltransferase 1 |
| AngII | angiotensin II |
| CAD | coronary artery disease |
| ECM | extracellular matrix |
| GPR19 | G protein-coupled receptor 19 |
| HAEC | human aortic endothelial cell |
| HASMC | human aortic smooth muscle cell |
| HUVEC | human umbilical vein endothelial cell |
| ICAM1 | intercellular adhesion molecule 1 |
| MMP | matrix metalloproteinase |
| NCEH | neutral cholesterol ester hydrolase |
| NFκB | nuclear factor κB |
| NOS | nitric oxide synthase |
| LDL | low-density lipoprotein |
| SRA | scavenger receptor class A |
| TNFα | tumor necrosis factor α |
| VCAM1 | vascular cell adhesion molecule 1 |
| VSMC | vascular smooth muscle cell |
References
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Paoli, F.D.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage phenotypes and their modulation in atherosclerosis. Circ. J. 2014, 78, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.; Trevaskis, J.L.; Lam, D.D.; Sutton, G.M.; Koza, R.A.; Chouljenko, V.N.; Kousoulas, K.G.; Rogers, P.M.; Kesterson, R.A.; Thearle, M.; et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 2008, 8, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.M.; Yosten, G.L.; Samson, W.K. Adropin acts in brain to inhibit water drinking: Potential interaction with the orphan G protein-coupled receptor, GPR19. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R476–R480. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Al-Omran, M.; Teoh, H.; Verma, S. Adropin is a novel regulator of endothelial function. Circulation 2010, 122, S185–S192. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Kuloglu, T.; Aydin, S.; Eren, M.N.; Yilmaz, M.; Kalayci, M.; Sahin, I.; Kocaman, N.; Citil, C.; Kendir, Y. Expression of adropin in rat brain, cerebellum, kidneys, heart, liver, and pancreas in streptozotocin-induced diabetes. Mol. Cell. Biochem. 2013, 380, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; DeMars, K.M.; Hawkins, K.E.; Candelario-Jalil, E. Adropin reduces paracellular permeability of rat brain endothelial cells exposed to ischemia-like conditions. Peptides 2016, 81, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lu, Z.; Burchell, S.; Nowrangi, D.; Manaenko, A.; Li, X.; Xu, Y.; Xu, N.; Tang, J.; Dai, H.; et al. Adropin preserves the blood-brain barrier through a Notch1/Hes1 pathway after intracerebral hemorrhage in mice. J. Neurochem. 2017, 143, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Zhao, P.; Wu, M.C.; Liu, J.; Yin, W. Serum adropin levels are decreased in patients with acute myocardial infarction. Regul. Pept. 2014, 190–191, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, L.; Xu, W.; Li, J.; Wang, B.; Gu, X.; Chen, J. Correlation of serum adropin level with coronary artery disease. Zhonghua Yi Xue Za Zhi 2014, 94, 1255–1257. [Google Scholar] [PubMed]
- Zhao, L.P.; Xu, W.T.; Wang, L.; You, T.; Chan, S.P.; Zhao, X.; Yang, X.J. Serum adropin level in patients with stable coronary artery disease. Heart Lung Circ. 2015, 24, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fang, J.; Chen, L.; Zhao, Z.; Luo, Y.; Lin, C.; Fan, L. Low serum adropin is associated with coronary atherosclerosis in type 2 diabetic and non-diabetic patients. Clin. Chem. Lab. Med. 2014, 52, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Beigi, A.; Shirzad, N.; Nikpour, F.; Nasli-Esfahani, E.; Emamgholipour, S.; Bandarian, F. Association between serum adropin levels and gestational diabetes mellitus; a case-control study. Gynecol. Endocrinol. 2015, 31, 939–941. [Google Scholar] [CrossRef] [PubMed]
- Yosaee, S.; Khodadost, M.; Esteghamati, A.; Speakman, J.R.; Shidfar, F.; Nazari, M.N.; Bitarafan, V.; Djafarian, K. Metabolic syndrome patients have lower levels of adropin when compared with healthy overweight/obese and lean subjects. Am. J. Men’s Health 2017, 11, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Altincik, A.; Sayin, O. Evaluation of the relationship between serum adropin levels and blood pressure in obese children. J. Pediatr. Endocrinol. Metab. 2015, 28, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Tam, C.S.; Stanhope, K.L.; Wolfe, B.M.; Ali, M.R.; O’Keeffe, M.; St-Onge, M.P.; Ravussin, E.; Havel, P.J. Low circulating adropin concentrations with obesity and aging correlate with risk factors for metabolic disease and increase after gastric bypass surgery in humans. J. Clin. Endocrinol. Metab. 2012, 97, 3783–3791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, L.; Yang, Y.J.; Ge, R.K.; Zhou, M.; Hu, H.; Liu, H.; Cui, J.; Li, L.L.; Dong, Y.F.; et al. Aerobic exercise improves endothelial function and serum adropin levels in obese adolescents independent of body weight loss. Sci. Rep. 2017, 7, 17717. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.P.; You, T.; Chan, S.P.; Chen, J.C.; Xu, W.T. Adropin is associated with hyperhomocysteine and coronary atherosclerosis. Exp. Ther. Med. 2016, 11, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Topuz, M.; Celik, A.; Aslantas, T.; Demir, A.K.; Aydin, S.; Aydin, S. Plasma adropin levels predict endothelial dysfunction like flow-mediated dilatation in patients with type 2 diabetes mellitus. J. Investig. Med. 2013, 61, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Oruc, C.U.; Akpinar, Y.E.; Dervisoglu, E.; Amikishiyev, S.; Salmaslıoglu, A.; Gurdol, F.; Omer, B. Low concentrations of adropin are associated with endothelial dysfunction as assessed by flow-mediated dilatation in patients with metabolic syndrome. Clin. Chem. Lab. Med. 2017, 55, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Rohwedder, I.; Montanez, E.; Beckmann, K.; Bengtsson, E.; Dunér, P.; Nilsson, J.; Soehnlein, O.; Fässler, R. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol. Med. 2012, 4, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Mugabe, B.E.; Yaghini, F.A.; Song, C.Y.; Buharalioglu, C.K.; Waters, C.M.; Malik, K.U. Angiotensin II-induced migration of vascular smooth muscle cells is mediated by p38 mitogen-activated protein kinase-activated c-Src through spleen tyrosine kinase and epidermal growth factor receptor transactivation. J. Pharmacol. Exp. Ther. 2010, 332, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Pakala, R.; Katagiri, T.; Benedict, C.R. Synergistic effect of urotensin II with mildly oxidized LDL on DNA synthesis in vascular smooth muscle cells. Circulation 2001, 104, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, T.Y.; Park, J.; Ha, H.; Kang, S.W.; Kim, Y.S. Effects of tautomycetin on proliferation and fibronectin secretion in vascular smooth muscle cells and glomerular mesangial cells. Transplant. Proc. 2005, 37, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.; Zhang, J.; Gao, S.; Rossi, J.; McGuinness, O.P.; Halem, H.H.; Culler, M.D.; Mynatt, R.L.; Butler, A.A. Adropin deficiency is associated with increased adiposity and insulin resistance. Obesity 2012, 20, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zeng, K.; Liu, Q.; Guo, Z.; Zhang, S.; Chen, X.; Lin, J.; Wen, J.; Zhao, C.; Lin, X.; et al. Adropin deficiency worsens HFD-induced metabolic defects. Cell Death Dis. 2017, 8, e3008. [Google Scholar] [CrossRef] [PubMed]
- Akcılar, R.; Koçak, F.E.; Şimşek, H.; Akcılar, A.; Bayat, Z.; Ece, E.; Kökdaşgil, H. Antidiabetic and hypolipidemic effects of adropinin streoptozotocin-induced type 2 diabetic rats. Bratisl. Lek. Listy 2016, 117, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Akcılar, R.; Koçak, F.E.; Şimşek, H.; Akcılar, A.; Bayat, Z.; Ece, E.; Kökdaşgil, H. The effect of adropin on lipid and glucose metabolism in rats with hyperlipidemia. Iran. J. Basic Med. Sci. 2016, 19, 245–251. [Google Scholar] [PubMed]
- Gao, S.; McMillan, R.P.; Zhu, Q.; Lopaschuk, G.D.; Hulver, M.W.; Butler, A.A. Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol. Metab. 2015, 4, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Li, H.; Zhu, X.; Gu, H.; Chen, J.; Wang, L.; Harding, P.; Xu, W. Inverse correlation between plasma adropin and ET-1 levels in essential hypertension: A cross-sectional study. Medicine 2015, 94, e1712. [Google Scholar] [CrossRef] [PubMed]
- Çelik, H.T.; Akkaya, N.; Erdamar, H.; Gok, S.; Kazanci, F.; Demircelik, B.; Cakmak, M.; Yigitoglu, R. The effects of valsartan and amlodipine on the levels of irisin, adropin, and perilipin. Clin. Lab. 2015, 61, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Gulen, B.; Eken, C.; Kucukdagli, O.T.; Serinken, M.; Kocyigit, A.; Kılıc, E.; Uyarel, H. Adropin levels and target organ damage secondary to high blood pressure in the ED. Am. J. Emerg. Med. 2016, 34, 2061–2064. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ouyang, F.; Zhou, S. Adropin as a novel energy factor likely has the ability to regulate blood pressure. Med. Hypotheses 2015, 85, 234. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Kathiresan, S. Genetics of coronary artery disease: Discovery, biology and clinical translation. Nat. Rev. Genet. 2017, 18, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Amos, C.I.; Lee, A.T.; Lu, E.; Remmers, E.F.; Kastner, D.L.; Seldin, M.F.; Criswell, L.A.; Plenge, R.M.; Holers, V.M.; et al. REL, a member of the NF-κB family of transcription factor, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 2009, 41, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Fujie, S.; Hasegawa, N.; Kurihara, T.; Sanada, K.; Hamaoka, T.; Iemitsu, M. Association between aerobic exercise training effects of serum adropin level, arterial stiffness, and adiposity in obese elderly adults. Appl. Physiol. Nutr. Metab. 2017, 42, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; St-Onge, M.P.; Siebert, E.A.; Medici, V.; Stanhope, K.L.; Havel, P.J. Differential responses of plasma adropin concentrations to dietary glucose or fluctose consumption in humans. Sci. Rep. 2015, 5, 14691. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Watanabe, R.; Konii, H.; Shirai, R.; Sato, K.; Matsuyama, T.; Ishibashi-Ueda, H.; Koba, S.; Kobayashi, Y.; Hirano, T.; et al. Counteractive effects of omentin-1 against atherogenesis. Cardiovasc. Res. 2016, 110, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Watanabe, H.; Takahashi, Y.; Kojima, M.; Konii, H.; Watanabe, K.; Shirai, R.; Sato, K.; Matsuyama, T.; Ishibashi-Ueda, H.; et al. Atheroprotective effects of tumor necrosis factor-stimulated gene-6. JACC Basic Transl. Sci. 2016, 1, 496–509. [Google Scholar] [CrossRef]
- Shirai, R.; Sato, K.; Yamashita, T.; Yamaguchi, M.; Okano, T.; Watanabe-Kominato, K.; Watanabe, R.; Matsuyama, T.; Ishibashi-Ueda, H.; Koba, S.; et al. Neopterin counters vascular inflammation and atherosclerosis. J. Am. Heart Assoc. 2018, 7, e007359. [Google Scholar] [CrossRef] [PubMed]
- Leosco, D.; Fineschi, M.; Pierli, C.; Fiaschi, A.; Ferrara, N.; Bianco, S.; Longobardi, G.; Pisani, E.; Bravi, A.; Rengo, F. Intracoronary serotonin release after high-pressure coronary stenting. Am. J. Cardiol. 1999, 84, 1317–1322. [Google Scholar] [CrossRef]
- Schuijt, M.P.; van Kats, J.P.; de Zeeuw, S.; Duncker, D.J.; Verdouw, P.D.; Schalekamp, M.A.D.H.; Danser, A.H.J. Cardiac interstitial fluid levels of angiotensin I and II in the pig. J. Hypertens. 1999, 17, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Ozawa, N.; Mori, Y.; Takahashi, Y.; Watanabe-Kominato, K.; Shirai, R.; Watanabe, R.; Sato, K.; Matsuyama, T.; Ishibashi-Ueda, H.; et al. Catestatin prevents macrophage-driven atherosclerosis but not arterial injury-induced neointimal hyperplasia. Thromb. Haemost. 2018, 118, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Watanabe, R.; Sato, Y.; Ozawa, N.; Kojima, M.; Watanabe-Kominato, K.; Shirai, R.; Sato, K.; Hirano, T.; Watanabe, T. Novel phytopeptide osmotin mimics preventive effects of adiponectin on vascular inflammation and atherosclerosis. Metabolism 2018, 83, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Shirai, R.; Shinooka, R.; Hasegawa, A.; Kichise, T.; Koba, S.; Matsuyama, T.; Kobayashi, Y.; Ishibashi-Ueda, H.; Watanabe, T. Potent vasoconstrictor kisspeptin-10 induces atherosclerotic plaque progression and instability: Reversal by its receptor GPR54 antagonist. J. Am. Heart Assoc. 2017, 6, e005790. [Google Scholar] [CrossRef] [PubMed]
- Naito, C.; Hashimoto, M.; Watanabe, K.; Shirai, R.; Takahashi, Y.; Kojima, M.; Watanabe, R.; Sato, K.; Iso, Y.; Matsuyama, T.; et al. Facilitatory effects of fetuin-A on atherosclerosis. Atherosclerosis 2016, 246, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, A.; Sato, K.; Shirai, R.; Watanabe, R.; Yamamoto, K.; Watanabe, K.; Nohtomi, K.; Hirano, T.; Watanabe, T. Vasoprotective effects of urocortin 1 against atherosclerosis in vitro and in vivo. PLoS ONE 2014, 9, e0110866. [Google Scholar] [CrossRef] [PubMed]
- Konii, H.; Sato, K.; Kikuchi, S.; Okiyama, H.; Watanabe, R.; Hasegawa, A.; Yamamoto, K.; Itoh, F.; Hirano, T.; Watanabe, T. Stimulatory effects of cardiotrophin 1 on atherosclerosis. Hypertension 2013, 62, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nishio, K.; Kanome, T.; Matsuyama, T.; Koba, S.; Sakai, T.; Sato, K.; Hongo, S.; Nose, K.; Ota, H.; et al. Impact of salusin-α and -β on human macrophage foam cell formation and coronary atherosclerosis. Circulation 2008, 117, 638–648. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 17 Weeks Old | 21 Weeks Old | ||
|---|---|---|---|---|
| Before | Adropin | Adropin | Adropin | |
| 0 μg/kg/h | 5 μg/kg/h | 10 μg/kg/h | ||
| N | 6 | 6 | 5 | 5 |
| Body weight (g) | 26.3 ± 0.7 | 28.5 ± 0.6 * | 29.0 ± 0.6 * | 28.7 ± 0.4 * |
| Food intake (g/day) | 3.9 ± 0.5 | 4.1 ± 0.3 | 4.0 ± 0.1 | 4.2 ± 0.5 |
| Systolic blood pressure (mm Hg) | 92.3 ± 1.1 | 92.7 ± 1.7 | 92.2 ± 2.4 | 94.6 ± 1.8 |
| Diastolic blood pressure (mm Hg) | 70.4 ± 1.4 | 72.0 ± 2.0 | 69.9 ± 2.4 | 70.4 ± 0.5 |
| Total cholesterol (mg/dL) | 2136.7 ± 149.9 | 2224.0 ± 183.6 | 1845.3 ± 56.7 | 2170.0 ± 138.7 |
| Triglyceride (mg/dL) | 285.4 ± 52.7 | 295.1 ± 41.7 | 300.9 ± 46.0 | 308.3 ± 99.5 |
| Free fatty acid (mEq/L) | 4.2 ± 0.6 | 2.7 ± 0.9 | 2.6 ± 0.4 | 2.0 ± 0.8 |
| Glucose (mg/dL) | 258.6 ± 34.2 | 261.3 ± 33.9 | 248.8 ± 21.3 | 255.5 ± 24.1 |
| Insulin (pmol/L) | 30.1 ± 7.6 | 42.7 ± 20.8 | 76.4 ± 8.8 | 72.4 ± 45.7 |
| HOMA-IR | 2.8 ± 0.8 | 3.4 ± 1.4 | 6.6 ± 1.0 | 7.1 ± 5.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, K.; Yamashita, T.; Shirai, R.; Shibata, K.; Okano, T.; Yamaguchi, M.; Mori, Y.; Hirano, T.; Watanabe, T. Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 1293. https://doi.org/10.3390/ijms19051293
Sato K, Yamashita T, Shirai R, Shibata K, Okano T, Yamaguchi M, Mori Y, Hirano T, Watanabe T. Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation. International Journal of Molecular Sciences. 2018; 19(5):1293. https://doi.org/10.3390/ijms19051293
Chicago/Turabian StyleSato, Kengo, Tomoyuki Yamashita, Remina Shirai, Koichiro Shibata, Taisuke Okano, Maho Yamaguchi, Yusaku Mori, Tsutomu Hirano, and Takuya Watanabe. 2018. "Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation" International Journal of Molecular Sciences 19, no. 5: 1293. https://doi.org/10.3390/ijms19051293
APA StyleSato, K., Yamashita, T., Shirai, R., Shibata, K., Okano, T., Yamaguchi, M., Mori, Y., Hirano, T., & Watanabe, T. (2018). Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation. International Journal of Molecular Sciences, 19(5), 1293. https://doi.org/10.3390/ijms19051293