An SCFFBXO28 E3 Ligase Protects Pancreatic β-Cells from Apoptosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
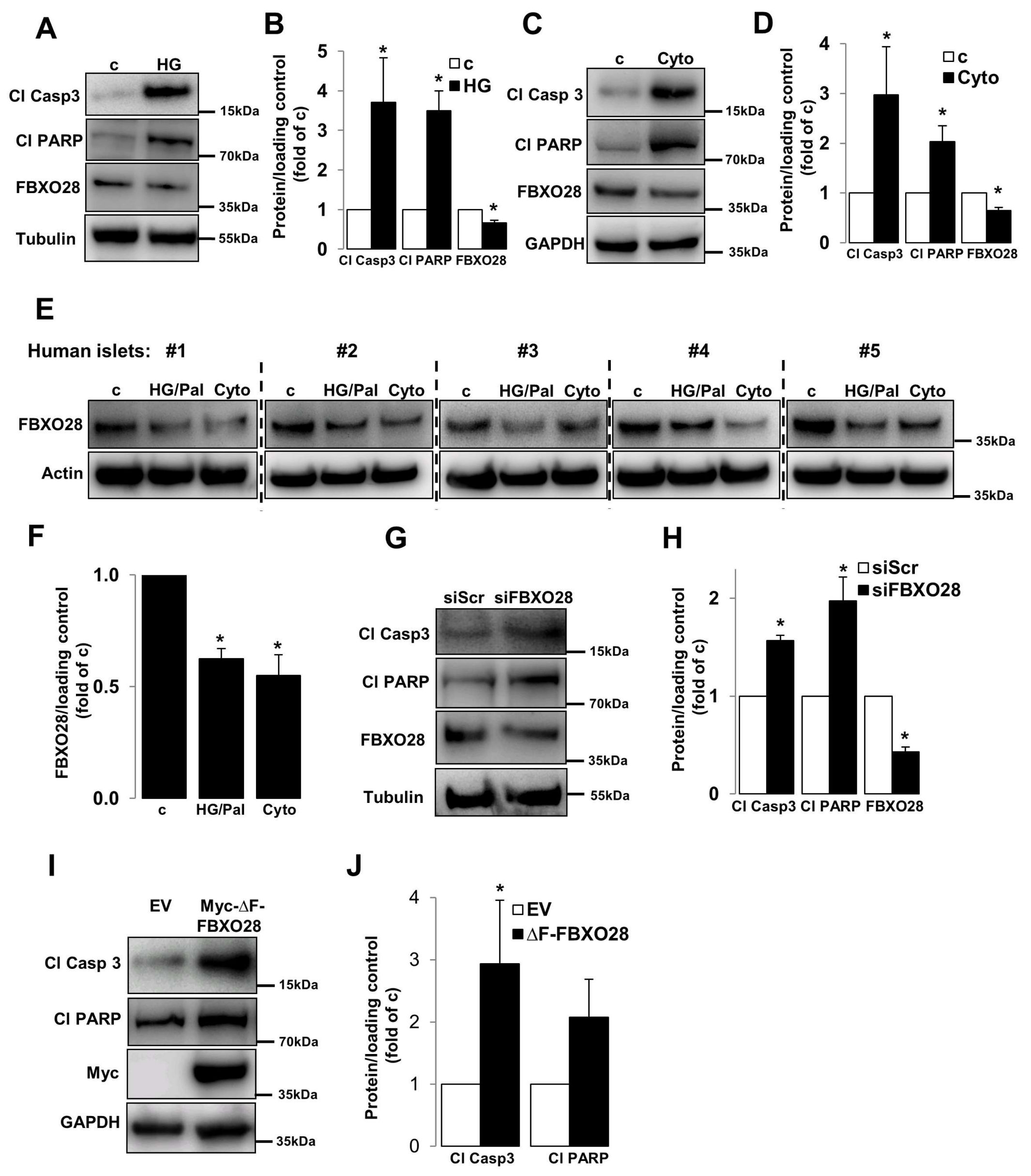
2.1. Loss of FBXO28 Induces β-Cell Apoptosis
2.2. Overexpression of FBXO28 Protects β-Cells from Apoptosis
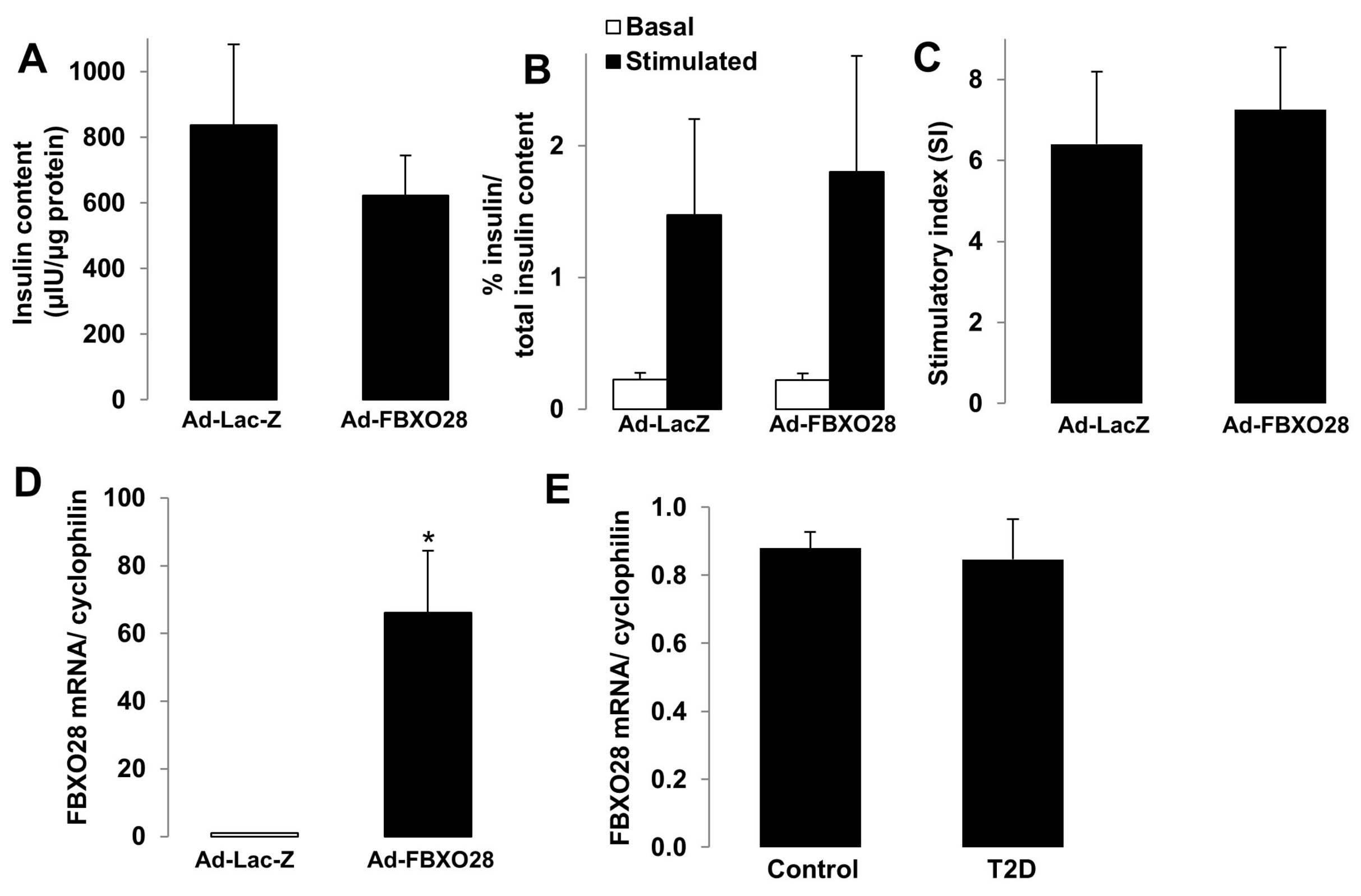
2.3. FBXO28 Does Not Regulate β-Cell Function
2.4. FBXO28 Overexpression Does Not Improve β-Cell Function in T2D Human Islets
3. Materials and Methods
3.1. Islet Isolation, Cell Culture, and Treatment
3.2. Transfections
3.3. Adenoviral Infection
3.4. Western Blot Analysis
3.5. Glucose-Stimulated Insulin Secretion
3.6. RNA Extraction and RT-PCR Analysis
3.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kurrer, M.O.; Pakala, S.V.; Hanson, H.L.; Katz, J.D. β cell apoptosis in T cell-mediated autoimmune diabetes. Proc. Natl. Acad. Sci. USA 1997, 94, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of β cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Roel, C.; Tanwir, M.; Wills, R.; Perianayagam, N.; Wang, P.; Fiaschi-Taesch, N.M. Definition of a Skp2-c-Myc Pathway to Expand Human B-cells. Sci. Rep. 2016, 6, 28461. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.E.; Kay, T.W. Intracellular pathways of pancreatic β-cell apoptosis in type 1 diabetes. Diabetes Metab. Res. Rev. 2011, 27, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Β-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced β-cell production of interleukin-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Gregg, B.; Blandino-Rosano, M.; Cras-Meneur, C.; Bernal-Mizrachi, E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol. Aspects Med. 2015, 42, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Lupse, B.; Kido, Y.; Leibowitz, G.; Maedler, K. mTORC1 Signaling: A Double-Edged Sword in Diabetic β Cells. Cell Metab. 2018, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosc. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Bugliani, M.; Liechti, R.; Cheon, H.; Suleiman, M.; Marselli, L.; Kirkpatrick, C.; Filipponi, F.; Boggi, U.; Xenarios, I.; Syed, F.; et al. Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin-proteasome system in pancreatic β cell dysfunction. Mol. Cell. Endocrinol. 2013, 367, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Litwak, S.A.; Wali, J.A.; Pappas, E.G.; Saadi, H.; Stanley, W.J.; Varanasi, L.C.; Kay, T.W.; Thomas, H.E.; Gurzov, E.N. Lipotoxic Stress Induces Pancreatic β-Cell Apoptosis through Modulation of Bcl-2 Proteins by the Ubiquitin-Proteasome System. J. Diabetes Res. 2015, 2015, 280615. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Gurlo, T.; Rivera, J.F.; Butler, P.C. UCHL1 deficiency exacerbates human islet amyloid polypeptide toxicity in β-cells: Evidence of interplay between the ubiquitin/proteasome system and autophagy. Autophagy 2014, 10, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Broca, C.; Varin, E.; Armanet, M.; Tourrel-Cuzin, C.; Bosco, D.; Dalle, S.; Wojtusciszyn, A. Proteasome dysfunction mediates high glucose-induced apoptosis in rodent β cells and human islets. PLoS ONE 2014, 9, e92066. [Google Scholar]
- Hofmeister-Brix, A.; Lenzen, S.; Baltrusch, S. The ubiquitin-proteasome system regulates the stability and activity of the glucose sensor glucokinase in pancreatic β-cells. Biochem. J. 2013, 456, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Huang, C.J.; Gurlo, T.; Daval, M.; Matveyenko, A.V.; Rizza, R.A.; Butler, A.E.; Butler, P.C. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 2011, 60, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Hartley, T.; Brumell, J.; Volchuk, A. Emerging roles for the ubiquitin-proteasome system and autophagy in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1–E10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaniuk, N.A.; Kiraly, M.; Bates, H.; Vranic, M.; Volchuk, A.; Brumell, J.H. Ubiquitinated-protein aggregates form in pancreatic β-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007, 56, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Villamil, M.A.; Liang, Q.; Chen, J.; Choi, Y.S.; Hou, S.; Lee, K.H.; Zhuang, Z. Serine phosphorylation is critical for the activation of ubiquitin-specific protease 1 and its interaction with WD40-repeat protein UAF1. Biochemistry 2012, 51, 9112–9123. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, D.; Ng, H.F.; Sharifi, H.R.; Mahmoudi, S.; Cerrato, V.S.; Fredlund, E.; Magnusson, K.; Nilsson, H.; Malyukova, A.; Rantala, J.; et al. CDK-mediated activation of the SCFFBXO28 ubiquitin ligase promotes MYC-driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Mol. Med. 2013, 5, 1067–1086. [Google Scholar] [CrossRef] [PubMed]
- Kratz, A.S.; Richter, K.T.; Schlosser, Y.T.; Schmitt, M.; Shumilov, A.; Delecluse, H.J.; Hoffmann, I. Fbxo28 promotes mitotic progression and regulates topoisomerase IIα-dependent DNA decatenation. Cell cycle 2016, 15, 3419–3431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Naya, F.J.; Huang, H.P.; Qiu, Y.; Mutoh, H.; DeMayo, F.J.; Leiter, A.B.; Tsai, M.J. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in β2/neuroD-deficient mice. Genes. Dev. 1997, 11, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.P.; Chu, K.; Nemoz-Gaillard, E.; Elberg, D.; Tsai, M.J. Neogenesis of β-cells in adult β2/NeuroD-deficient mice. Mol. Endocrinol. 2002, 16, 541–551. [Google Scholar] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic β-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Docherty, H.M.; Hay, C.W.; Ferguson, L.A.; Barrow, J.; Durward, E.; Docherty, K. Relative contribution of PDX-1, MafA and E47/β2 to the regulation of the human insulin promoter. Biochem. J. 2005, 389 Pt 3, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, F.T.; Paroni, F.; Sauter, N.S.; Shu, L.; Ribaux, P.; Haataja, L.; Strieter, R.M.; Oberholzer, J.; King, C.C.; Maedler, K. CXCL10 impairs β cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009, 9, 125–139. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorrepati, K.D.D.; He, W.; Lupse, B.; Yuan, T.; Maedler, K.; Ardestani, A. An SCFFBXO28 E3 Ligase Protects Pancreatic β-Cells from Apoptosis. Int. J. Mol. Sci. 2018, 19, 975. https://doi.org/10.3390/ijms19040975
Gorrepati KDD, He W, Lupse B, Yuan T, Maedler K, Ardestani A. An SCFFBXO28 E3 Ligase Protects Pancreatic β-Cells from Apoptosis. International Journal of Molecular Sciences. 2018; 19(4):975. https://doi.org/10.3390/ijms19040975
Chicago/Turabian StyleGorrepati, Kanaka Durga Devi, Wei He, Blaz Lupse, Ting Yuan, Kathrin Maedler, and Amin Ardestani. 2018. "An SCFFBXO28 E3 Ligase Protects Pancreatic β-Cells from Apoptosis" International Journal of Molecular Sciences 19, no. 4: 975. https://doi.org/10.3390/ijms19040975
APA StyleGorrepati, K. D. D., He, W., Lupse, B., Yuan, T., Maedler, K., & Ardestani, A. (2018). An SCFFBXO28 E3 Ligase Protects Pancreatic β-Cells from Apoptosis. International Journal of Molecular Sciences, 19(4), 975. https://doi.org/10.3390/ijms19040975