A Whole Germline BRCA2 Gene Deletion: How to Learn from CNV In Silico Analysis

 , , ,
, , ,
Abstract
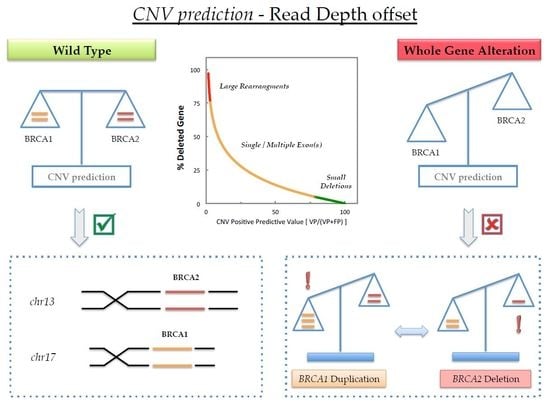
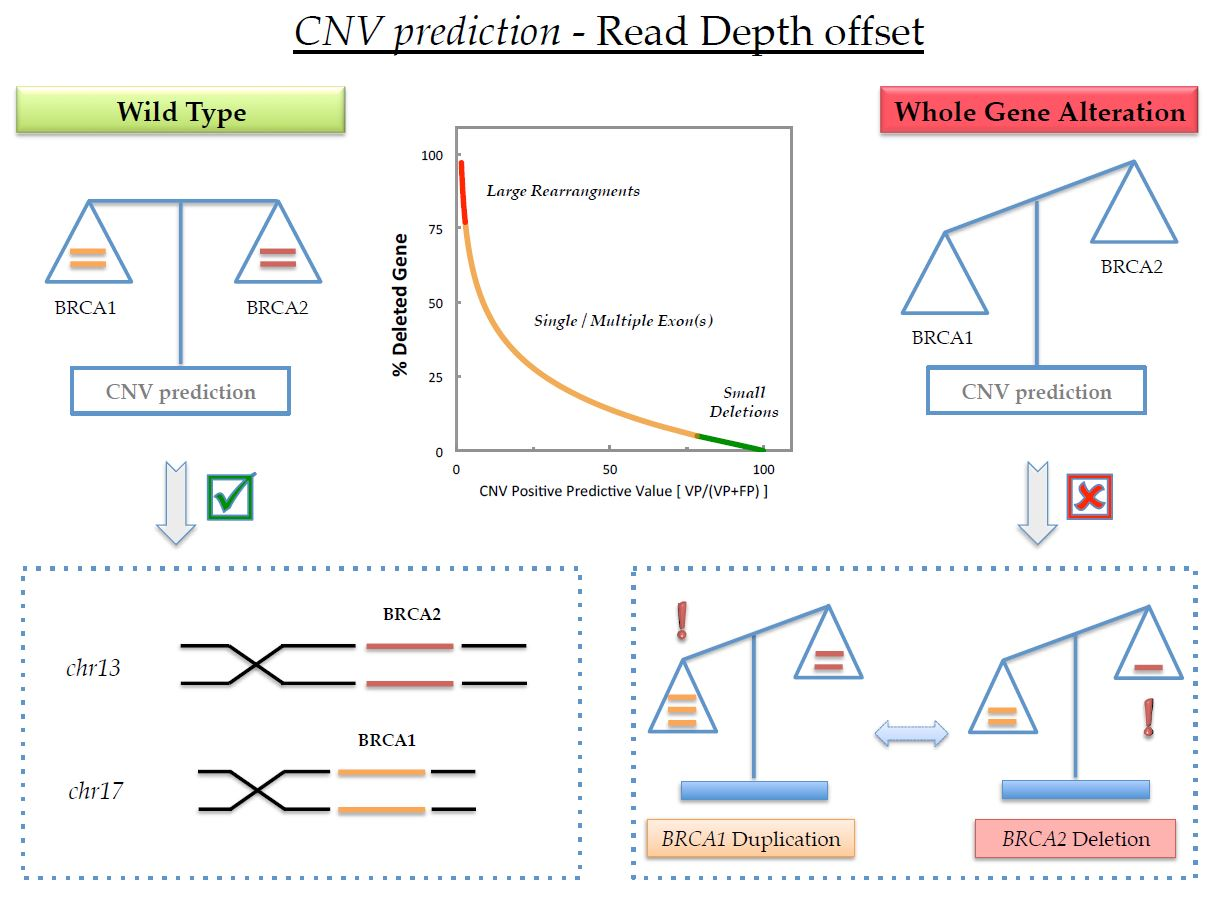
1. Introduction
2. Results
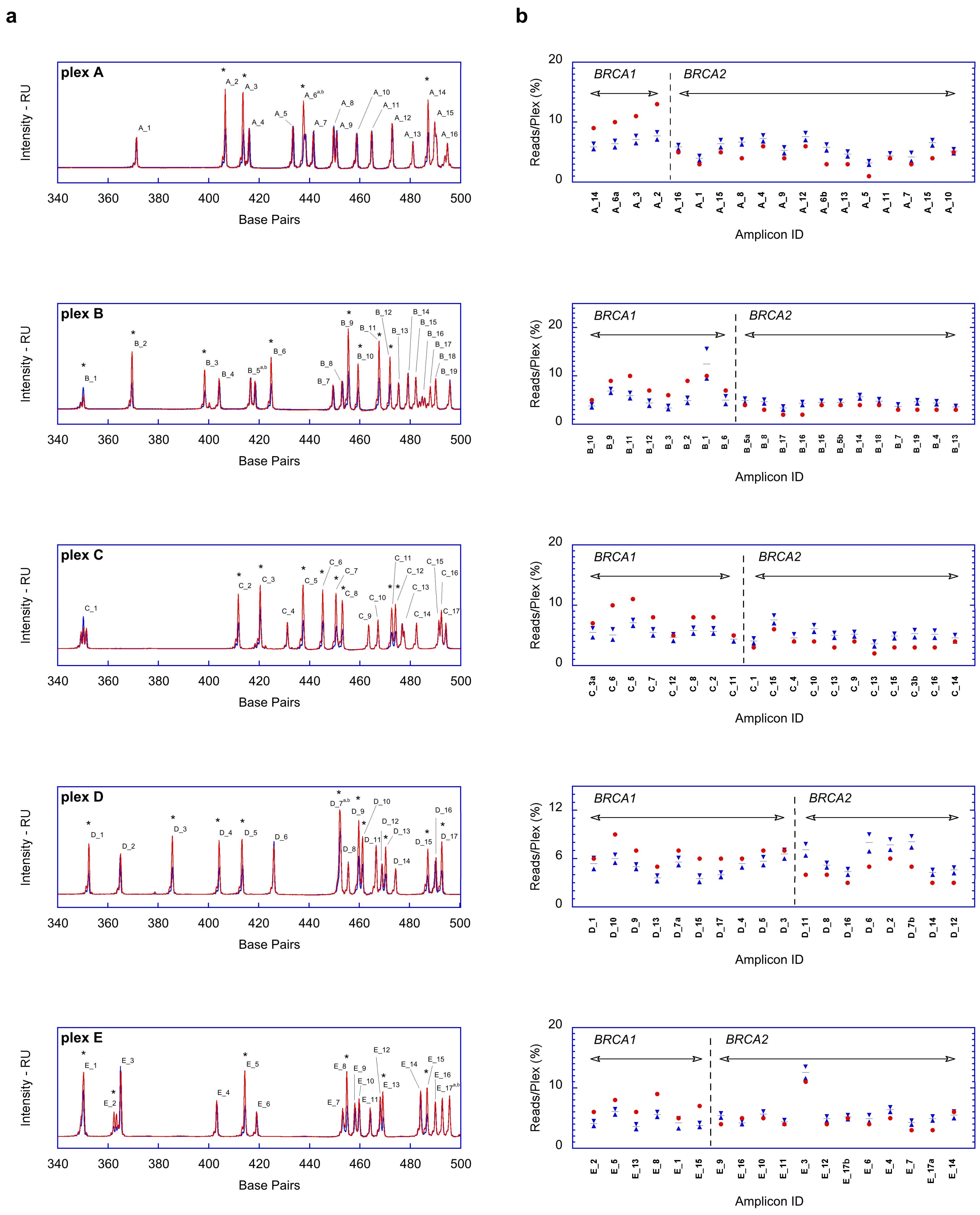
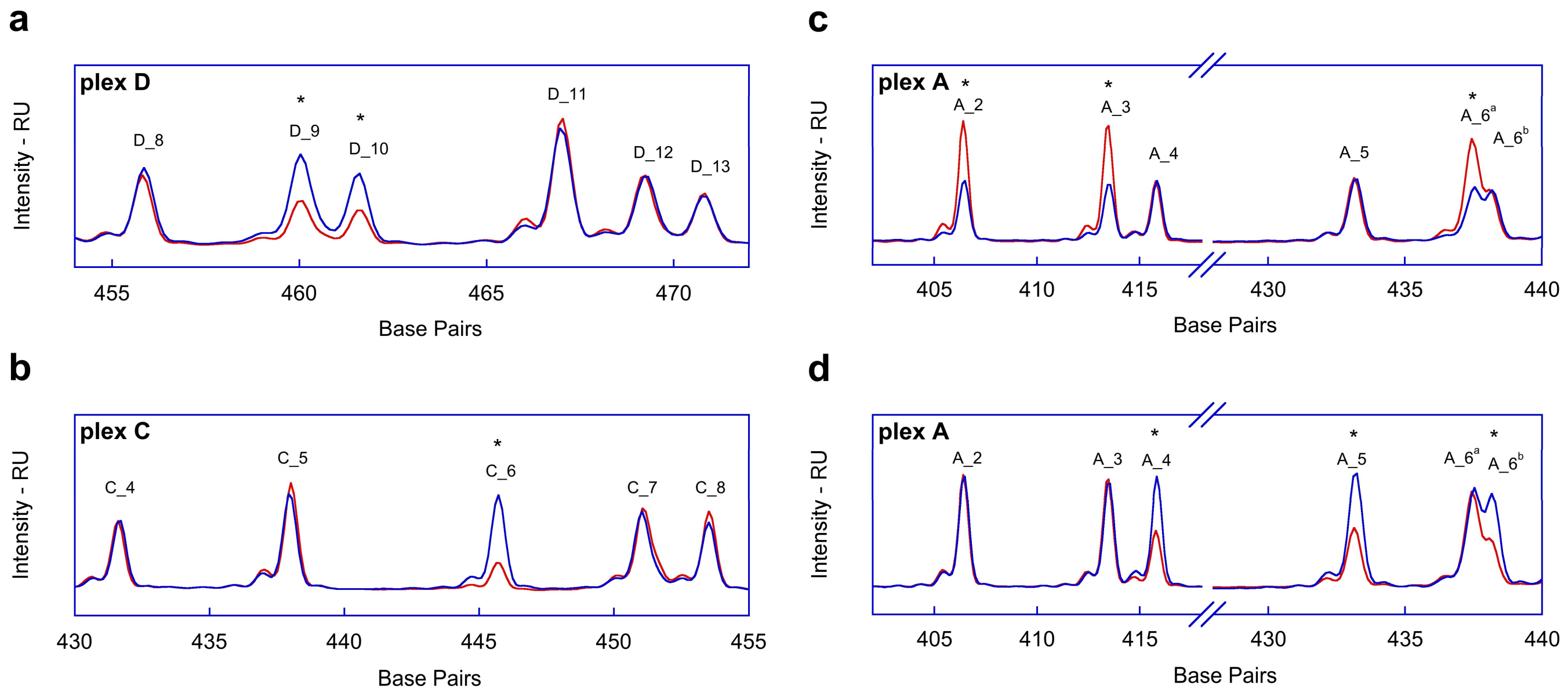
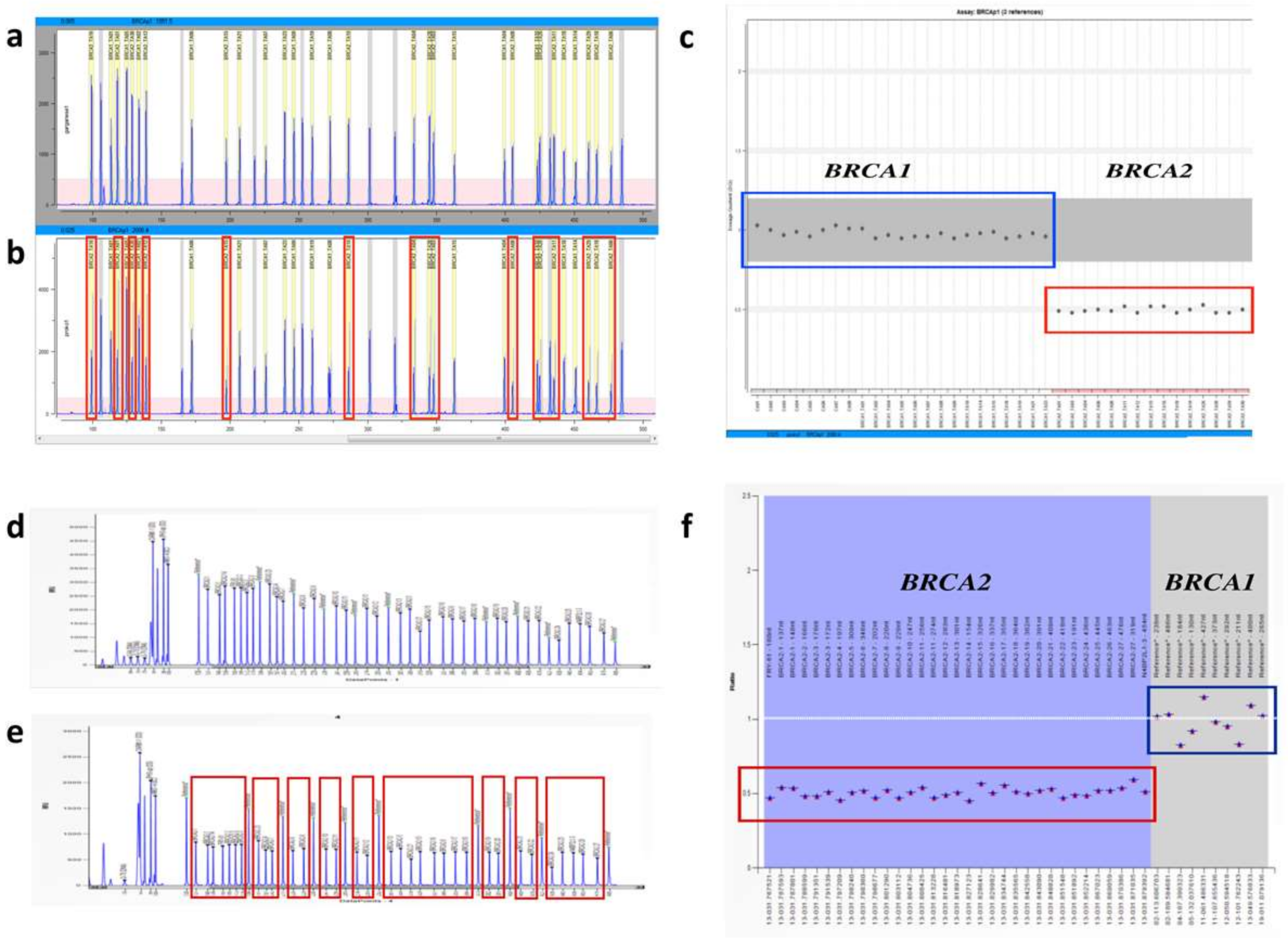
2.1. Fragment Analysis (FA)
2.2. MPS Analysis
2.3. MAQ Analysis
2.4. MLPA Analysis
3. Discussion
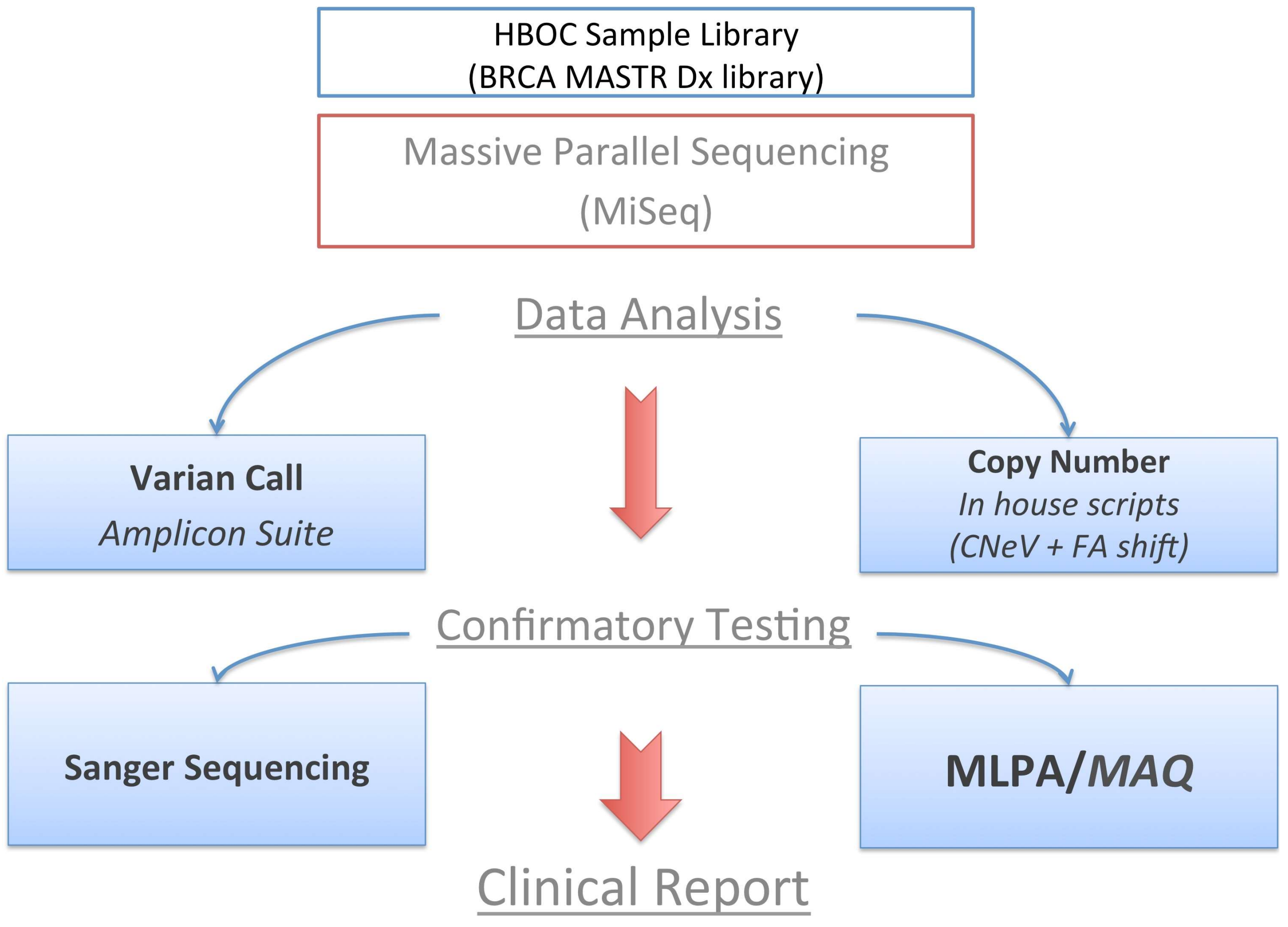
4. Materials and Methods
4.1. DNA Samples
4.2. MPS Analysis
4.3. MAQ Analysis
4.4. MLPA Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
Abbreviations
| BRCA1/2 | BReast CAncer susceptibility genes 1/2 |
| CNV | Copy Number Variant |
| DDM | Data-Driven Medicine |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| HBOC | Hereditary Breast and Ovarian Syndrome |
| MLPA | Multiplex Ligation-dependent Probe Amplification |
| MAQ | Multiplex Amplicon Quantification |
| MPS | Massively Parallel Sequencing |
| NGS | Next Generation Sequencing |
| PGM | Personal Genome Medicine |
| VBA | Visual Basic for Applications |
| WGS | Whole Genome Sequencing |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plex A | Plex B | Plex C | Plex D | Plex E | |||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Amplicon | ID | Amplicon | ID | Amplicon | ID | Amplicon | ID | Amplicon |
| A_1 | BRCA2_ex09_01 | B_1 | BRCA1_ex20_01 | C_1 | BRCA2_ex06_01 | D_1 | BRCA1_ex03_01 | E_1 | BRCA1_ex22_01 |
| A_2 | BRCA1_ex23_01 | B_2 | BRCA1_ex19_01 | C_2 | BRCA1_ex14_01 | D_2 | BRCA2_ex11_13 | E_2 | BRCA1_ex09_01 |
| A_3 | BRCA1_ex11_12 | B_3 | BRCA1_ex16_02 | C_3a | BRCA1_ex02_01 | D_3 | BRCA1_ex17_01 | E_3 | BRCA2_ex11_14 |
| A_4 | BRCA2_ex11_07 | B_4 | BRCA2_ex25_02 | C_3b | BRCA2_ex19_01 | D_4 | BRCA1_ex13_01 | E_4 | BRCA2_ex21_01 |
| A_5 | BRCA2_ex13_01 | B_5a | BRCA2_ex02_01 | C_4 | BRCA2_ex10_04 | D_5 | BRCA1_ex16_01 | E_5 | BRCA1_ex10_01 |
| A_6a | BRCA1_ex11_07 | B_5b | BRCA2_ex11_09 | C_5 | BRCA1_ex11_05 | D_6 | BRCA2_ex11_11 | E_6 | BRCA2_ex12_01 |
| A_6b | BRCA2_ex11_19 | B_6 | BRCA1_ex21_01 | C_6 | BRCA1_ex06_01 | D_7a | BRCA1_ex11_06 | E_7 | BRCA2_ex22_01 |
| A_7 | BRCA2_ex15_01 | B_7 | BRCA2_ex16_01 | C_7 | BRCA1_ex11_08 | D_7b | BRCA2_ex14_02 | E_8 | BRCA1_ex11_10 |
| A_8 | BRCA2_ex11_01 | B_8 | BRCA2_ex08_01 | C_8 | BRCA1_ex12_01 | D_8 | BRCA2_ex10_03 | E_9 | BRCA2_ex03_02 |
| A_9 | BRCA2_ex11_10 | B_9 | BRCA1_ex11_01 | C_9 | BRCA2_ex11_12 | D_9 | BRCA1_ex05_01 | E_10 | BRCA2_ex11_02 |
| A_10 | BRCA2_ex24_01 | B_10 | BRCA1_ex08_01 | C_10 | BRCA2_ex11_05 | D_10 | BRCA1_ex07_01 | E_11 | BRCA2_ex11_06 |
| A_11 | BRCA2_ex14_01 | B_11 | BRCA1_ex11_14 | C_11 | BRCA1_ex18_01 | D_11 | BRCA2_ex05_01 | E_12 | BRCA2_ex11_17 |
| A_12 | BRCA2_ex11_15 | B_12 | BRCA1_ex15_01 | C_12 | BRCA1_ex11_13 | D_12 | BRCA2_ex27_03 | E_13 | BRCA1_ex11_02 |
| A_13 | BRCA2_ex11_21 | B_13 | BRCA2_ex27_02 | C_13a | BRCA2_ex11_08 | D_13 | BRCA1_ex11_03 | E_14 | BRCA2_ex27_01 |
| A_14 | BRCA1_ex11_04 | B_14 | BRCA2_ex11_18 | C_13b | BRCA2_ex11_16 | D_14 | BRCA2_ex25_01 | E_15 | BRCA1_ex24_01 |
| A_15a | BRCA2_ex10_01 | B_15 | BRCA2_ex11_04 | C_14 | BRCA2_ex23_01 | D_15 | BRCA1_ex11_09 | E_16 | BRCA2_ex04_01 |
| A_15b | BRCA2_ex18_01 | B_16 | BRCA2_ex10_05 | C_15a | BRCA2_ex18_02 | D_16 | BRCA2_ex11_03 | E_17a | BRCA2_ex11_22 |
| A_16 | BRCA2_ex03_01 | B_17 | BRCA2_ex10_02 | C_15b | BRCA2_ex07_01 | D_17 | BRCA1_ex11_11 | E_17b | BRCA2_ex26_01 |
| B_18 | BRCA2_ex11_20 | C_16 | BRCA2_ex20_01 | ||||||
| B_19 | BRCA2_ex17_01 | ||||||||
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Infromation Core (BIC). National Human Research Institute. Available online: http://research.nhgri.nih.gov/bic/ (accessed on 5 October 2017).
- Concolino, P.; Mello, E.; Minucci, A.; Santonocito, C.; Scambia, G.; Giardina, B.; Capoluongo, E. Advanced tools for BRCA1/2 mutational screening: Comparison between two methods for large genomic rearrangements (LGRs) detection. Clin. Chem. Lab. Med. 2014, 52, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Minucci, A.; Scambia, G.; Santonocito, C.; Concolino, P.; Canu, G.; Mignone, F.; Saggese, I.; Guarino, D.; Costella, A.; Molinario, R.; et al. Clinical impact on ovarian cancer patients of massive parallel sequencing for BRCA mutation detection: The experience at Gemelli hospital and a literature review. Expert Rev. Mol. Diagn. 2015, 15, 1383–1403. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-associated hereditary breast and ovarian cancer. In Genereviews(r); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mefford, H.C., Stephens, K., Amemiya, A., Ledbetter, N., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Specifications for the MiSeq System. Available online: https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html (accessed on 10 January 2018).
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [PubMed]
- Agilent. MAQ USER Guide v1.0. Available online: https://www.agilent.com/en/products/next-generation-sequencing/amplicon-target-amplification-(multiplicom)/cancer-genetics/brca-maq (accessed on 7 February 2018).
- Kumps, C.; Van Roy, N.; Heyrman, L.; Goossens, D.; Del-Favero, J.; Noguera, R.; Vandesompele, J.; Speleman, F.; De Preter, K. Multiplex amplicon quantification (MAQ), a fast and efficient method for the simultaneous detection of copy number alterations in neuroblastoma. BMC Genom. 2010, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Nunziato, M.; Starnone, F.; Lombardo, B.; Pensabene, M.; Condello, C.; Verdesca, F.; Carlomagno, C.; De Placido, S.; Pastore, L.; Salvatore, F.; et al. Fast detection of a BRCA2 large genomic duplication by next generation sequencing as a single procedure: A case report. Int. J. Mol. Sci. 2017, 18, 2487. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Bella, M.A.; Capoluongo, E.; Carrera, P.; Clemente, C.; Colombo, N.; Cortesi, L.; De Rosa, G.; Fenizia, F.; Genuardi, M.; et al. Recommendations for the implementation of BRCA testing in the care and treatment pathways of ovarian cancer patients. Future Oncol. 2016, 12, 2071–2075. [Google Scholar] [CrossRef] [PubMed]
- Capoluongo, E. BRCA to the future: Towards best testing practice in the era of personalised healthcare. Eur. J. Hum. Genet. 2016, 24, S1–S2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Badoer, C.; Garrec, C.; Goossens, D.; Ellison, G.; Mills, J.; Dzial, M.; El Housni, H.; Berwouts, S.; Concolino, P.; Guibert-Le Guevellou, V.; et al. Performance of multiplicom’s BRCA MASTR Dx kit on the detection of BRCA1 and BRCA2 mutations in fresh frozen ovarian and breast tumor samples. Oncotarget 2016, 7, 81357–81366. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, D.; Scambia, G.; Pignata, S.; Sorio, R.; Amadio, G.; Lepori, S.; Mosconi, A.; Pisano, C.; Mangili, G.; Maltese, G.; et al. Prospective phase II trial of trabectedin in BRCA-mutated and/or BRCAness phenotype recurrent ovarian cancer patients: The MITO 15 trial. Ann. Oncol. 2016, 27, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Capoluongo, E.; Ellison, G.; López-Guerrero, J.A.; Penault-Llorca, F.; Ligtenberg, M.J.L.; Banerjee, S.; Singer, C.; Friedman, E.; Markiefka, B.; Schirmacher, P.; et al. Guidance statement on BRCA1/2 tumor testing in ovarian cancer patients. Semin. Oncol. 2017, 44, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Vetro, A.; Goidin, D.; Lesende, I.; Limongelli, I.; Ranzani, G.N.; Novara, F.; Bonaglia, M.C.; Rinaldi, B.; Franchi, F.; Manolakos, E.; et al. Diagnostic application of a capture based NGS test for the concurrent detection of variants in sequence and copy number as well as LOH. Clin. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Concolino, P.; Rizza, R.; Hackmann, K.; Minucci, A.; Scaglione, G.L.; De Bonis, M.; Costella, A.; Zuppi, C.; Schrock, E.; Capoluongo, E. Identification and characterization of a NEW BRCA2 rearrangement in an italian family with hereditary breast and ovarian cancer syndrome. Mol. Diagn. Ther. 2017, 21, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Concolino, P.; Rizza, R.; Hackmann, K.; Paris, I.; Minucci, A.; De Paolis, E.; Scambia, G.; Zuppi, C.; Schrock, E.; Capoluongo, E. Characterization of a new BRCA1 rearrangement in an italian woman with hereditary breast and ovarian cancer syndrome. Breast Cancer Res. Treat. 2017, 164, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.J. New challenges for BRCA testing: A view from the diagnostic laboratory. Eur. J. Hum. Genet. 2016, 24, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Feliubadalo, L.; Lopez-Doriga, A.; Castellsague, E.; del Valle, J.; Menendez, M.; Tornero, E.; Montes, E.; Cuesta, R.; Gomez, C.; Campos, O.; et al. Next-generation sequencing meets genetic diagnostics: Development of a comprehensive workflow for the analysis of BRCA1 and BRCA2 genes. Eur. J. Hum. Genet. 2013, 21, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef] [PubMed]
- Zare, F.; Dow, M.; Monteleone, N.; Hosny, A.; Nabavi, S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinform. 2017, 18, 286. [Google Scholar] [CrossRef] [PubMed]
- Molparia, B.; Nichani, E.; Torkamani, A. Assessment of circulating copy number variant detection for cancer screening. PLoS ONE 2017, 12, e0180647. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Purshouse, K.; Schuh, A.; Fairfax, B.P.; Knight, S.; Antoniou, P.; Dreau, H.; Popitsch, N.; Gatter, K.; Roberts, I.; Browning, L.; et al. Whole-genome sequencing identifies homozygous BRCA2 deletion guiding treatment in dedifferentiated prostate cancer. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001362. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Marchetti, C.; De Leo, R.; Musella, A.; Capoluongo, E.; Paris, I.; Benedetti Panici, P.; Scambia, G.; Fagotti, A. BRCA mutational status, initial disease presentation, and clinical outcome in high-grade serous advanced ovarian cancer: A multicenter study. Am. J. Obstet. Gynecol. 2017, 217, 334.e1–334.e9. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scaglione, G.L.; Concolino, P.; De Bonis, M.; De Paolis, E.; Minucci, A.; Ferrandina, G.; Scambia, G.; Capoluongo, E. A Whole Germline BRCA2 Gene Deletion: How to Learn from CNV In Silico Analysis. Int. J. Mol. Sci. 2018, 19, 961. https://doi.org/10.3390/ijms19040961
Scaglione GL, Concolino P, De Bonis M, De Paolis E, Minucci A, Ferrandina G, Scambia G, Capoluongo E. A Whole Germline BRCA2 Gene Deletion: How to Learn from CNV In Silico Analysis. International Journal of Molecular Sciences. 2018; 19(4):961. https://doi.org/10.3390/ijms19040961
Chicago/Turabian StyleScaglione, Giovanni Luca, Paola Concolino, Maria De Bonis, Elisa De Paolis, Angelo Minucci, Gabriella Ferrandina, Giovanni Scambia, and Ettore Capoluongo. 2018. "A Whole Germline BRCA2 Gene Deletion: How to Learn from CNV In Silico Analysis" International Journal of Molecular Sciences 19, no. 4: 961. https://doi.org/10.3390/ijms19040961
APA StyleScaglione, G. L., Concolino, P., De Bonis, M., De Paolis, E., Minucci, A., Ferrandina, G., Scambia, G., & Capoluongo, E. (2018). A Whole Germline BRCA2 Gene Deletion: How to Learn from CNV In Silico Analysis. International Journal of Molecular Sciences, 19(4), 961. https://doi.org/10.3390/ijms19040961