Role of Inflammation in Diabetic Retinopathy

Abstract
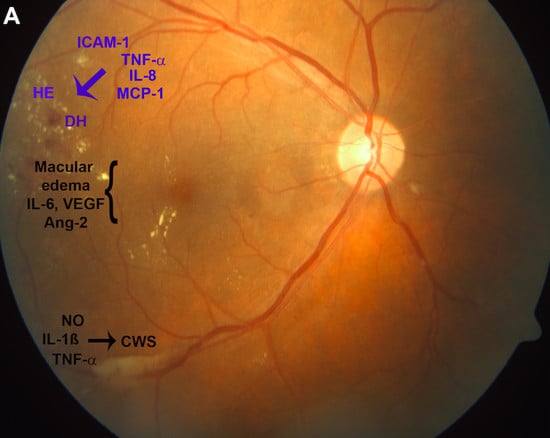
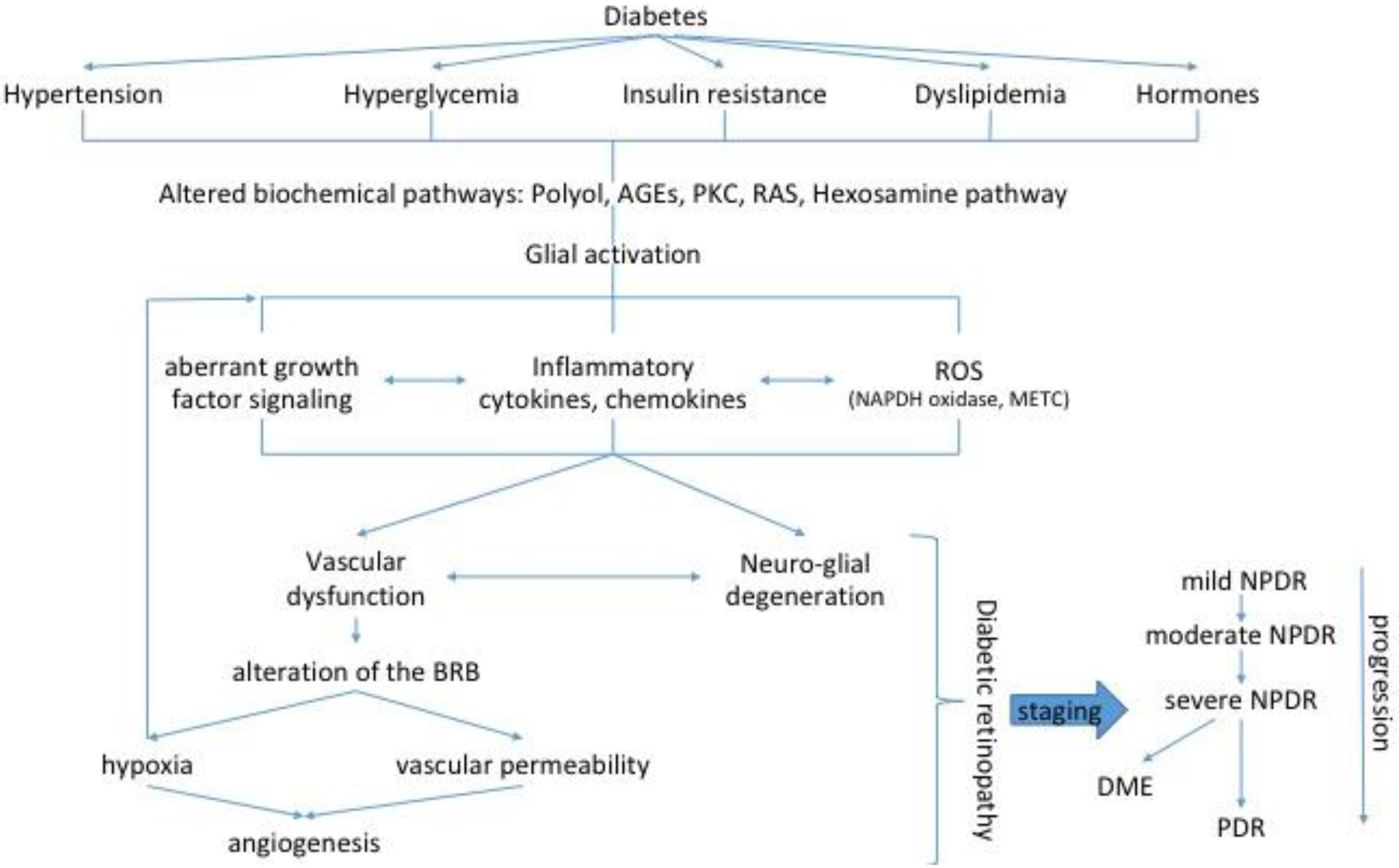
1. Introduction
2. Emergence of the Neurovascular Unit and Implications for Early DR
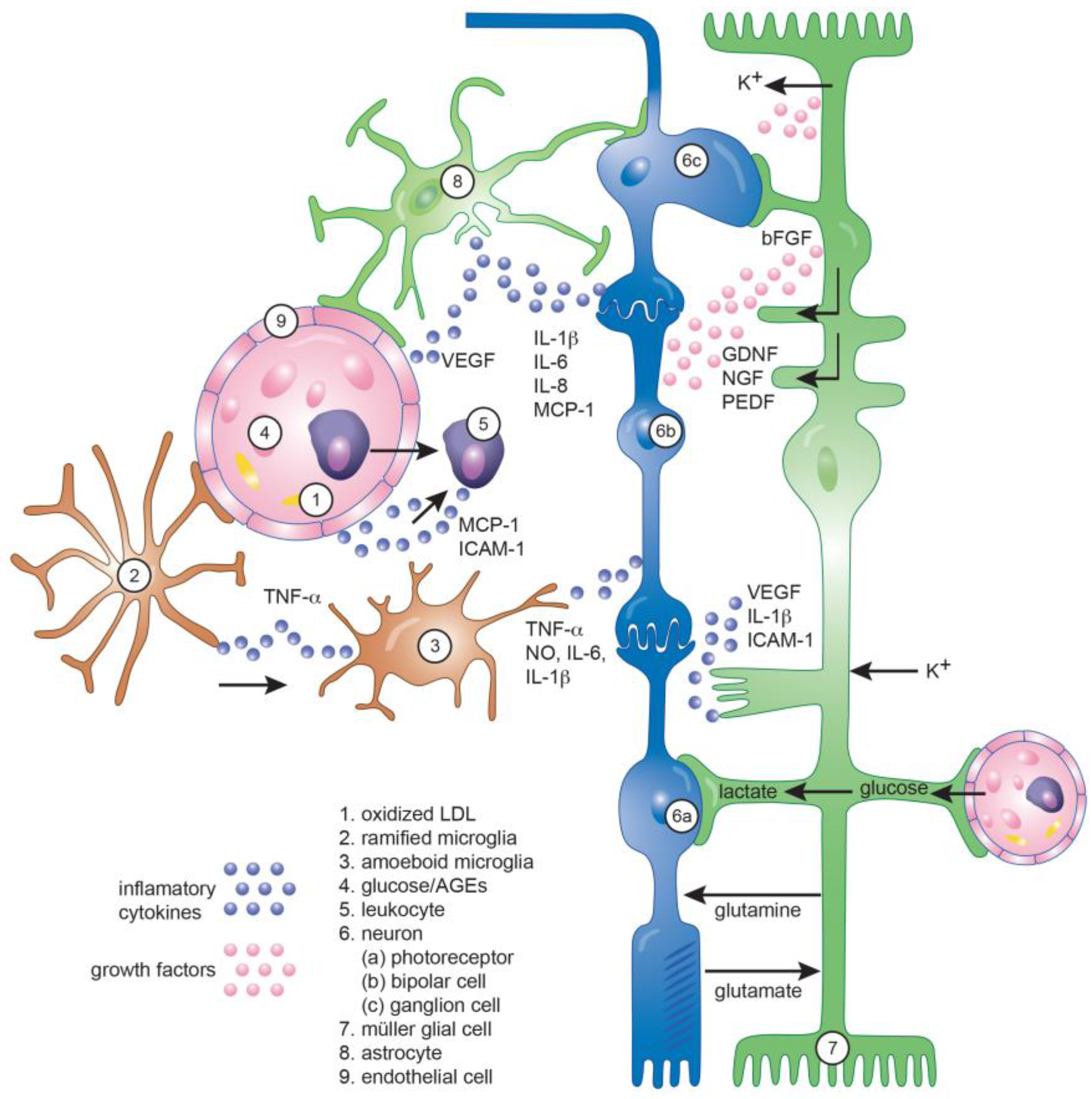
3. Role of Inflammation in Diabetic Retinopathy
3.1. Inflammation
3.2. Evidences of Inflammation in Diabetic Retinopathy Pathogenesis
3.3. Contribution to Vascular Pathology
3.4. Contribution to Neurodegeneration
4. Role of Glial Cells in Diabetic Retinopathy
4.1. Microglia
4.2. Müller Glial Cells
4.3. Astrocytes
4.4. Role of Inflammation in the Neuro-Glial Crosstalk in DR
5. Emerging Trends in Inflammation-Related Mediators of Diabetic Retinopathy
5.1. Alpha-Crystallins
5.2. Matrix Metalloproteinases
5.3. Toll-Like Receptors
5.4. Complement System
6. Therapeutic Concepts in Diabetic Retinopathy
6.1. Current Therapies
6.2. Inflammation-Targeting Therapies under Development and Validation
6.2.1. Nonsteroidal Anti-Inflammatory Drugs (NSAID)
6.2.2. Blocking Inflammatory Molecules
6.2.3. Inflammatory Growth Factors
6.2.4. The Renin-Angiotensin System (RAS)
6.2.5. Tetracyclines
6.2.6. Photobiomodulation
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGE | Advanced Glycation End-product |
| AH | Aqueous Humor |
| Ang | Angiopoietin |
| AT1R | Angiotensin II Receptor Type 1 |
| AQP | Aquaporin |
| BDNF | Brain-derived Neurotrophic factor |
| bFGF | Basic Fibroblast Growth Factor |
| BRB | Blood-retinal Barrier |
| CCL | C-C motif ligand |
| CCR | C-C receptor |
| CD | Cluster of differentiation |
| CNS | Central Nervous System |
| CNTF | Ciliary Neurotrophic Factor |
| COX | Cyclooxygenase |
| CRP | C-reactive Protein |
| CXCL | C-X-C motif ligand |
| DR | Diabetic Retinopathy |
| DCCT | Diabetes Control and Complications Trial |
| DME | Diabetic Macular Edema |
| EGF | Epidermal Growth factor |
| ERG | Electroretinogram |
| ERK | Extracellular Signal-Regulated Kinase |
| FDA | US Food and Drug Administration |
| GDNF | Glial-derived Neurotrophic factor |
| GFAP | Glial Fibrillary Acidic Protein |
| GM-CSF | Granulocyte Macrophage Colony-Stimulating Factor |
| HGF | Hepatocyte Growth Factor |
| HIF | Hypoxia-inducible Factor |
| HMGB | High Mobility Group Box |
| ICAM | Intracellular Adhesion Molecule |
| IGF | Insulin-like Growth Factor |
| IFN | Interferon |
| IL | Interleukin |
| iNOS | Inducible Nitrous Oxide Synthetase |
| IP | Interferon γ-induced Protein |
| IRBP | Interphotoreceptor Retinoid Binding Protein |
| IRMA | Intraretinal Microvascular Abnormality |
| IVTA | Intravitreal Triamcinolone Acetonide |
| LIF | Leukemia inhibitory factor |
| LPS | Lipopolysaccharide |
| MCP | Monocyte Chemoattractant Protein |
| MIP | Macrophage Inflammatory Protein |
| MMP | Matrix Metalloproteinase |
| NFG | Nerve Growth Factor |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric Oxide |
| NPDR | Non-proliferative Diabetic Retinopathy |
| NSAID | Nonsteroidal Anti-Inflammatory Drug |
| PAMP | Pathogen-associated Molecular Pattern |
| PDGF | Platelet-derived Growth Factor |
| PDR | Proliferative Diabetic Retinopathy |
| PEDF | Pigment Epithelium-derived Factor |
| PGE | Prostaglandin |
| PGF | Placental growth factor |
| PKC | Protein Kinase C |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PPV | Pars Plana Vitrectomy |
| RAS | Renin-Angiotensin System |
| RAGE | Receptor for Advanced Glycation End-product |
| RANTES | Regulated and normal T cell expressed and secreted |
| RGC | Retinal Ganglion Cell |
| ROS | Reactive Oxygen Species |
| RPE | Retinal Pigment Epithelium |
| SDF | Stromal-derived Factor |
| sTNF-R | Soluble Tumor Necrosis Factor-Receptor |
| STZ | Streptozotocin |
| TGF | Transforming Growth Factor |
| TLR | Toll-like Receptor |
| TNF | Tumor Necrosis Factor |
| UKPDS | United Kingdom Prospective Diabetes Study |
| VEGF | Vascular Endothelial Growth Factor |
References
- Klein, B.E. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol. 2007, 14, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Kempen, J.H.; O’Colmain, B.J.; Leske, M.C.; Haffner, S.M.; Klein, R.; Moss, S.E.; Taylor, H.R.; Hamman, R.F.; Eye Diseases Prevalence Research Group. The prevalence of diabetic retinopathy among adults in the United States. Arch. Ophthalmol. 2004, 122, 552–563. [Google Scholar] [PubMed]
- Zhang, X.; Saaddine, J.B.; Chou, C.F.; Cotch, M.F.; Cheng, Y.J.; Geiss, L.S.; Gregg, E.W.; Albright, A.L.; Klein, B.E.; Klein, R. Prevalence of diabetic retinopathy in the United States, 2005–2008. JAMA 2010, 304, 649–656. [Google Scholar] [CrossRef] [PubMed]
- AAO PPP Retina/Vitreous Panel, Hoskins Center for Quality Eye Care. Diabetic Retinopathy Ppp—Updated. 2016. Available online: http://www.aao.org/preferred-practice-pattern/diabetic-retinopathy-ppp--2014 (accessed on 30 October 2017).
- Frey, T.; Antonetti, D.A. Alterations to the blood-retinal barrier in diabetes: Cytokines and reactive oxygen species. Antioxid. Redox Signal 2011, 15, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zeng, H.; Bao, S.; Wang, N.; Gillies, M.C. Diabetic macular edema: New concepts in patho-physiology and treatment. Cell Biosci. 2014, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Daruich, A.; Matet, A.; Moulin, A.; Kowalczuk, L.; Nicolas, M.; Sellam, A.; Rothschild, P.R.; Omri, S.; Gelize, E.; Jonet, L.; et al. Mechanisms of macular edema: Beyond the surface. Prog. Retin. Eye Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Adamis, A.P.; Shima, D.T.; D’Amore, P.A.; Moulton, R.S.; O’Reilly, M.S.; Folkman, J.; Dvorak, H.F.; Brown, L.F.; Berse, B.; et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 1994, 145, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.A.; Kermany, D.S.; Waters, J.; Jansen, M.E.; Tyler, L. Diabetic macular edema: It is more than just VEGF. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Abcouwer, S.F.; Barber, A.J.; Jackson, G.R. An integrated approach to diabetic retinopathy research. Arch. Ophthalmol. 2011, 129, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Noma, H.; Mimura, T.; Eguchi, S.; Hori, S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 2009, 116, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Elner, S.G.; Elner, V.M.; Jaffe, G.J.; Stuart, A.; Kunkel, S.L.; Strieter, R.M. Cytokines in proliferative diabetic retinopathy and proliferative vitreoretinopathy. Curr. Eye Res. 1995, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Takeuchi, S.; Matsuda, A.; Tagawa, Y.; Mizue, Y.; Nishihira, J. Monocyte chemotactic protein-1 in the vitreous of patients with proliferative diabetic retinopathy. Ophthalmologica 2001, 215, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, Y.; Arora, S.; Erb-Downward, J.R.; McDonald, R.A.; Toews, G.B.; Huffnagle, G.B. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J. Immunol. 2005, 174, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Struyf, S.; Kangave, D.; Geboes, K.; Van Damme, J. Chemokines in proliferative diabetic retinopathy and proliferative vitreoretinopathy. Eur. Cytokine Netw. 2006, 17, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, D.; Wakabayashi, Y.; Usui, Y.; Okunuki, Y.; Kezuka, T.; Goto, H. Correlation of complement fragment C5a with inflammatory cytokines in the vitreous of patients with proliferative diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, Y.; Usui, Y.; Okunuki, Y.; Kezuka, T.; Takeuchi, M.; Goto, H.; Iwasaki, T. Correlation of vascular endothelial growth factor with chemokines in the vitreous in diabetic retinopathy. Retina 2010, 30, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Murata, T.; Tsujikawa, A.; Kirchhof, B.; Bursell, S.E.; Adamis, A.P. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am. J. Pathol. 2001, 158, 147–152. [Google Scholar] [CrossRef]
- Powell, E.D.; Field, R.A. Diabetic retinopathy and rheumatoid arthritis. Lancet 1964, 2, 17–18. [Google Scholar] [CrossRef]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and Caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, M.; Gerhardinger, C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia 2001, 44, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; Gardner, T.W.; Barber, A.J.; Antonetti, D.A.; Penn State Retina Research, G. Retinal neurodegeneration: Early pathology in diabetes. Clin. Exp. Ophthalmol. 2000, 28, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Park, J.W.; Park, S.J.; Kim, K.Y.; Chung, J.W.; Chun, M.H.; Oh, S.J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Enzsoly, A.; Szabo, A.; Kantor, O.; David, C.; Szalay, P.; Szabo, K.; Szel, A.; Nemeth, J.; Lukats, A. Pathologic alterations of the outer retina in streptozotocin-induced diabetes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Ostroy, S.E.; Frede, S.M.; Wagner, E.F.; Gaitatzes, C.G.; Janle, E.M. Decreased rhodopsin regeneration in diabetic mouse eyes. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3905–3909. [Google Scholar]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Muller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Abu-El-Asrar, A.M.; Dralands, L.; Missotten, L.; Al-Jadaan, I.A.; Geboes, K. Expression of apoptosis markers in the retinas of human subjects with diabetes. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2760–2766. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Rani, P.K.; Raman, R.; Pal, S.S.; Laxmi, G.; Gupta, M.; Sahu, C.; Vaitheeswaran, K.; Sharma, T. Is neuronal dysfunction an early sign of diabetic retinopathy? Microperimetry and spectral domain optical coherence tomography (Sd-Oct) study in individuals with diabetes, but no diabetic retinopathy. Eye (London) 2009, 23, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Van der Torren, K.; van Lith, G. Oscillatory potentials in early diabetic retinopathy. Doc. Ophthalmol. 1989, 71, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Sokol, S.; Moskowitz, A.; Skarf, B.; Evans, R.; Molitch, M.; Senior, B. Contrast sensitivity in diabetics with and without background retinopathy. Arch. Ophthalmol. 1985, 103, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.S.; Gunkel, R.D.; Podgor, M.J. Color vision defects in early diabetic retinopathy. Arch. Ophthalmol. 1986, 104, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.K.; Lin, M.M.; Lammer, J.; Prager, S.; Sarangi, R.; Silva, P.S.; Aiello, L.P. Disorganization of the retinal inner layers as a predictor of visual acuity in eyes with center-involved diabetic macular edema. JAMA Ophthalmol. 2014, 132, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, S.; Tadayoni, R.; Erginay, A.; Massin, P.; Dupas, B. Correlation between ganglion cell layer thinning and poor visual function after resolution of diabetic macular edema. Investig. Ophthalmol. Vis. Sci. 2015, 56, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Lee, S.H.; Chung, H.; Kim, H.C. Association between photoreceptor integrity and visual outcome in diabetic macular edema. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Agemy, S.A.; Scripsema, N.K.; Shah, C.M.; Chui, T.; Garcia, P.M.; Lee, J.G.; Gentile, R.C.; Hsiao, Y.S.; Zhou, Q.; Ko, T.; et al. Retinal vascular perfusion density mapping using optical coherence tomography angiography in normals and diabetic retinopathy patients. Retina 2015, 35, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Kaur, H. The role of toll-like receptors in diabetes-induced inflammation: Implications for vascular complications. Curr. Diabetes Rep. 2012, 12, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Adamis, A.P. Is diabetic retinopathy an inflammatory disease? Br. J. Ophthalmol. 2002, 86, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 95103. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Nakao, S.; Ishida, S.; Ishibashi, T. Leukocyte adhesion molecules in diabetic retinopathy. J. Ophthalmol. 2012, 2012, 279037. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Micera, A.; Bini, S.; Berton, M.; Esposito, G.; Midena, E. Aqueous humor biomarkers of muller cell activation in diabetic eyes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Khalfaoui, T.; Lizard, G.; Ouertani-Meddeb, A. Adhesion molecules (ICAM-1 and VCAM-1) and diabetic retinopathy in type 2 diabetes. J. Mol. Histol. 2008, 39, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Micera, A.; Bini, S.; Berton, M.; Esposito, G.; Midena, E. Proteome analysis of retinal glia cells-related inflammatory cytokines in the aqueous humour of diabetic patients. Acta Ophthalmol. 2016, 94, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gerhardinger, C.; Dagher, Z.; Hoehn, T.; Lorenzi, M. Aspirin at low-intermediate concentrations protects retinal vessels in experimental diabetic retinopathy through non-platelet-mediated effects. Diabetes 2005, 54, 3418–3426. [Google Scholar] [CrossRef] [PubMed]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of neurotrophins and inflammatory mediators in vitreous of patients with diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5594–5603. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hwang, D.K.; Song, X.; Tao, Y. Association between aqueous cytokines and diabetic retinopathy stage. J. Ophthalmol. 2017, 2017, 9402198. [Google Scholar] [CrossRef] [PubMed]
- Demircan, N.; Safran, B.G.; Soylu, M.; Ozcan, A.A.; Sizmaz, S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye (London) 2006, 20, 1366–1369. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Yoshida, A.; Ishibashi, T.; Elner, S.G.; Elner, V.M. Role of MCP-1 and MIP-1alpha in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J. Leukoc. Biol. 2003, 73, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, S.; Vrzalova, J.; Sobotova, M.; Hecova, L.; Ricarova, R.; Topolcan, O. The measurement of intraocular biomarkers in various stages of proliferative diabetic retinopathy using multiplex xmap technology. J. Ophthalmol. 2015, 2015, 424783. [Google Scholar] [CrossRef] [PubMed]
- Doganay, S.; Evereklioglu, C.; Er, H.; Turkoz, Y.; Sevinc, A.; Mehmet, N.; Savli, H. Comparison of serum no, TNF-alpha, IL-1beta, SIL-2r, IL-6 and il-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye (London) 2002, 16, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.J.; Menicucci, G.; McGuire, P.G.; Das, A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab. Investig. 2005, 85, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.I.; Hykin, P.G.; Gregor, Z.J.; Boulton, M.; Cree, I.A. Angiopoietin concentrations in diabetic retinopathy. Br. J. Ophthalmol. 2005, 89, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Chalam, K.V.; Grover, S.; Sambhav, K.; Balaiya, S.; Murthy, R.K. Aqueous interleukin-6 levels are superior to vascular endothelial growth factor in predicting therapeutic response to bevacizumab in age-related macular degeneration. J. Ophthalmol. 2014, 2014, 502174. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Qin, W.; Kirchhof, B.; Mitsiades, N.; Wiegand, S.J.; Rudge, J.; Yancopoulos, G.D.; Adamis, A.P. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am. J. Pathol. 2002, 160, 501–509. [Google Scholar] [CrossRef]
- Rungger-Brandle, E.; Dosso, A.A.; Leuenberger, P.M. Glial reactivity, an early feature of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1971–1980. [Google Scholar]
- Cusick, M.; Chew, E.Y.; Chan, C.C.; Kruth, H.S.; Murphy, R.P.; Ferris, F.L., 3rd. Histopathology and regression of retinal hard exudates in diabetic retinopathy after reduction of elevated serum lipid levels. Ophthalmology 2003, 110, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gerhardinger, C.; Lorenzi, M. Early complement activation and decreased levels of glycosylphosphatidylinositol-anchored complement inhibitors in human and experimental diabetic retinopathy. Diabetes 2002, 51, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Bronson, S.K.; Freeman, W.M.; Gardner, T.W.; Jefferson, L.S.; Kester, M.; Kimball, S.R.; Krady, J.K.; LaNoue, K.F.; et al. Diabetic retinopathy: Seeing beyond glucose-induced microvascular disease. Diabetes 2006, 55, 2401–2411. [Google Scholar] [CrossRef] [PubMed]
- Cardona, S.M.; Mendiola, A.S.; Yang, Y.C.; Adkins, S.L.; Torres, V.; Cardona, A.E. Disruption of fractalkine signaling leads to microglial activation and neuronal damage in the diabetic retina. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, S.; Palacios, E.; Romacho, T.; Villalobos, L.; Peiro, C.; Sanchez-Ferrer, C.F. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2014, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, A.; Han, P.Y.; Nakagawa, T.; Johnson, R.J.; Grant, M.B.; Campbell-Thompson, M.; Jarajapu, Y.P.; Lei, B.; Hauswirth, W.W. Diabetic enos-knockout mice develop accelerated retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5240–5246. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Yamashita, H.; Sakata, K.; Noma, H.; Mimura, T.; Suzuki, M.; Eguchi, S.; Hori, S. Vitreous levels of vascular endothelial growth factor and intercellular adhesion molecule 1 are related to diabetic macular edema. Ophthalmology 2005, 112, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-White, J.L.; Glazer, L.; Downer, R.; Furge, K.; Boguslawski, E.; Duesbery, N.S. Identification of VEGF-independent cytokines in proliferative diabetic retinopathy vitreous. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6472–6480. [Google Scholar] [CrossRef] [PubMed]
- Adamiec-Mroczek, J.; Oficjalska-Mlynczak, J.; Misiuk-Hojlo, M. Proliferative diabetic retinopathy-the influence of diabetes control on the activation of the intraocular molecule system. Diabetes Res. Clin. Pract. 2009, 84, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Saxena, S.; Khanna, V.K.; Shukla, R.K.; Meyer, C.H. Status of serum VEGF and ICAM-1 and its association with external limiting membrane and inner segment-outer segment junction disruption in type 2 diabetes mellitus. Mol. Vis. 2013, 19, 1760–1768. [Google Scholar] [PubMed]
- Burgos, R.; Simo, R.; Audi, L.; Mateo, C.; Mesa, J.; Garcia-Ramirez, M.; Carrascosa, A. Vitreous levels of vascular endothelial growth factor are not influenced by its serum concentrations in diabetic retinopathy. Diabetologia 1997, 40, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Hang, H.; Yuan, S.; Yang, Q.; Yuan, D.; Liu, Q. Multiplex bead array assay of plasma cytokines in type 2 diabetes mellitus with diabetic retinopathy. Mol. Vis. 2014, 20, 1137–1145. [Google Scholar] [PubMed]
- Jonas, J.B.; Jonas, R.A.; Neumaier, M.; Findeisen, P. Cytokine concentration in aqueous humor of eyes with diabetic macular edema. Retina 2012, 32, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Ghodasra, D.H.; Fante, R.; Gardner, T.W.; Langue, M.; Niziol, L.M.; Besirli, C.; Cohen, S.R.; Dedania, V.S.; Demirci, H.; Jain, N.; et al. Safety and feasibility of quantitative multiplexed cytokine analysis from office-based vitreous aspiration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Yan, H. Roles of elevated intravitreal IL-1beta and IL-10 levels in proliferative diabetic retinopathy. Indian J. Ophthalmol. 2014, 62, 699–701. [Google Scholar] [PubMed]
- Kim, M.; Kim, Y.; Lee, S.J. Comparison of aqueous concentrations of angiogenic and inflammatory cytokines based on optical coherence tomography patterns of diabetic macular edema. Indian J. Ophthalmol. 2015, 63, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Kang, M.H.; Seong, M.; Cho, H.Y. Comparison of aqueous concentrations of angiogenic and inflammatory cytokines in diabetic macular oedema and macular oedema due to branch retinal vein occlusion. Br. J. Ophthalmol. 2012, 96, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Le-Ruppert, K.C. Activated t lymphocytes in epiretinal membranes from eyes of patients with proliferative diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 1995, 233, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Sato, T.; Tanaka, A.; Muraoka, T.; Taguchi, M.; Sakurai, Y.; Karasawa, Y.; Ito, M. Elevated levels of cytokines associated with TH2 and TH17 cells in vitreous fluid of proliferative diabetic retinopathy patients. PLoS ONE 2015, 10, e0137358. [Google Scholar] [CrossRef] [PubMed]
- Meleth, A.D.; Agron, E.; Chan, C.C.; Reed, G.F.; Arora, K.; Byrnes, G.; Csaky, K.G.; Ferris, F.L., 3rd; Chew, E.Y. Serum inflammatory markers in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4295–4301. [Google Scholar] [CrossRef] [PubMed]
- Noma, H.; Mimura, T.; Yasuda, K.; Motohashi, R.; Kotake, O.; Shimura, M. Aqueous humor levels of soluble vascular endothelial growth factor receptor and inflammatory factors in diabetic macular edema. Ophthalmologica 2017, 238, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; Nishikawa, M.; Nishimura, T.; Mitsuma, Y.; Matsumura, M. Unbalanced vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor in diabetic retinopathy. Am. J. Ophthalmol. 2002, 134, 348–353. [Google Scholar] [CrossRef]
- Duh, E.J.; Yang, H.S.; Haller, J.A.; De Juan, E.; Humayun, M.S.; Gehlbach, P.; Melia, M.; Pieramici, D.; Harlan, J.B.; Campochiaro, P.A.; et al. Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor: Implications for ocular angiogenesis. Am. J. Ophthalmol. 2004, 137, 668–674. [Google Scholar] [PubMed]
- Wang, S.; Gottlieb, J.L.; Sorenson, C.M.; Sheibani, N. Modulation of thrombospondin 1 and pigment epithelium-derived factor levels in vitreous fluid of patients with diabetes. Arch. Ophthalmol. 2009, 127, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Yamashita, H.; Nakamura, S.; Mimura, T.; Eguchi, S.; Noma, H.; Hori, S. Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2006, 113, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwickerath, R.; Pfeiffer, A.; Blum, W.F.; Freyberger, H.; Klein, M.; Losche, C.; Rollmann, R.; Schatz, H. Vitreous levels of the insulin-like growth factors I and II, and the insulin-like growth factor binding proteins 2 and 3, increase in neovascular eye disease. Studies in nondiabetic and diabetic subjects. J. Clin. Investig. 1993, 92, 2620–2625. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.; Russell, B.; Fitzgerald, C.; Merimee, T.J. Insulin-like growth factors in vitreous. Studies in control and diabetic subjects with neovascularization. Diabetes 1986, 35, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Sivalingam, A.; Kenney, J.; Brown, G.C.; Benson, W.E.; Donoso, L. Basic fibroblast growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch. Ophthalmol. 1990, 108, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Al Kahtani, E.; Xu, Z.; Al Rashaed, S.; Wu, L.; Mahale, A.; Tian, J.; Abboud, E.B.; Ghazi, N.G.; Kozak, I.; Gupta, V.; et al. Vitreous levels of placental growth factor correlate with activity of proliferative diabetic retinopathy and are not influenced by bevacizumab treatment. Eye (London) 2017, 31, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.; Barry, P.; Shaw, C.; Duffy, J.; Kennedy, S. Platelet derived growth factor and fibroblast growth factor basic levels in the vitreous of patients with vitreoretinal disorders. Br. J. Ophthalmol. 1998, 82, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Yngen, M.; Ostenson, C.G.; Hu, H.; Li, N.; Hjemdahl, P.; Wallen, N.H. Enhanced p-selectin expression and increased soluble cd40 ligand in patients with type 1 diabetes mellitus and microangiopathy: Evidence for platelet hyperactivity and chronic inflammation. Diabetologia 2004, 47, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Loukovaara, S.; Koivunen, P.; Ingles, M.; Escobar, J.; Vento, M.; Andersson, S. Elevated protein carbonyl and HIF-1alpha levels in eyes with proliferative diabetic retinopathy. Acta Ophthalmol. 2014, 92, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Wang, Y. Intravitreous vascular endothelial growth factor and hypoxia-inducible factor 1A in patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 2009, 148, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; McGuire, P.G.; Das, A. Diabetic retinopathy and inflammation: Novel therapeutic targets. Middle East Afr. J. Ophthalmol. 2012, 19, 52–59. [Google Scholar] [PubMed]
- Aplin, A.C.; Gelati, M.; Fogel, E.; Carnevale, E.; Nicosia, R.F. Angiopoietin-1 and vascular endothelial growth factor induce expression of inflammatory cytokines before angiogenesis. Physiol. Genom. 2006, 27, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Leali, D.; Dell’Era, P.; Stabile, H.; Sennino, B.; Chambers, A.F.; Naldini, A.; Sozzani, S.; Nico, B.; Ribatti, D.; Presta, M. Osteopontin (ETA-1) and fibroblast growth factor-2 cross-talk in angiogenesis. J. Immunol. 2003, 171, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Esselman, W.J.; Jump, D.B.; Busik, J.V. Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal vascular endothelial cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4342–4347. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simo, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Durham, J.T.; Herman, I.M. Microvascular modifications in diabetic retinopathy. Curr. Diabetes Rep. 2011, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.L. Pathogenesis of diabetic retinopathy. Diabetes 1989, 38, 1203–1206. [Google Scholar] [CrossRef] [PubMed]
- Dell’omo, R.; Semeraro, F.; Bamonte, G.; Cifariello, F.; Romano, M.R.; Costagliola, C. Vitreous mediators in retinal hypoxic diseases. Mediat. Inflamm. 2013, 2013, 935301. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, C.; Romano, V.; De Tollis, M.; Aceto, F.; dell’Omo, R.; Romano, M.R.; Pedicino, C.; Semeraro, F. TNF-alpha levels in tears: A novel biomarker to assess the degree of diabetic retinopathy. Mediat. Inflamm. 2013, 2013, 629529. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Yoshida, A.; Ishibashi, T. Induction of IL-8, MCP-1, and BFGF by TNF-alpha in retinal glial cells: Implications for retinal neovascularization during post-ischemic inflammation. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, C.; Sonoda, K.H.; Egashira, K.; Qiao, H.; Hisatomi, T.; Nakao, S.; Ishibashi, M.; Charo, I.F.; Sakamoto, T.; Murata, T.; et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J. Leukoc. Biol. 2003, 74, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, E.; Taguchi, H.; Anand, A.; Ambati, B.K.; Gragoudas, E.S.; Miller, J.W.; Adamis, A.P.; Ambati, J. Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2743–2749. [Google Scholar] [CrossRef]
- Sennlaub, F.; Valamanesh, F.; Vazquez-Tello, A.; El-Asrar, A.M.; Checchin, D.; Brault, S.; Gobeil, F.; Beauchamp, M.H.; Mwaikambo, B.; Courtois, Y.; et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation 2003, 108, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Ayalasomayajula, S.P.; Kompella, U.B. Celecoxib, a selective cyclooxygenase-2 inhibitor, inhibits retinal vascular endothelial growth factor expression and vascular leakage in a streptozotocin-induced diabetic rat model. Eur. J. Pharmacol. 2003, 458, 283–289. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Mohammad, G.; Nawaz, M.I.; Siddiquei, M.M. High-mobility group box-1 modulates the expression of inflammatory and angiogenic signaling pathways in diabetic retina. Curr. Eye Res. 2015, 40, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Fiuza, C.; Bustin, M.; Talwar, S.; Tropea, M.; Gerstenberger, E.; Shelhamer, J.H.; Suffredini, A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003, 101, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Treutiger, C.J.; Mullins, G.E.; Johansson, A.S.; Rouhiainen, A.; Rauvala, H.M.; Erlandsson-Harris, H.; Andersson, U.; Yang, H.; Tracey, K.J.; Andersson, J.; et al. High mobility group 1 b-box mediates activation of human endothelium. J. Intern. Med. 2003, 254, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Croll, S.D.; Ransohoff, R.M.; Cai, N.; Zhang, Q.; Martin, F.J.; Wei, T.; Kasselman, L.J.; Kintner, J.; Murphy, A.J.; Yancopoulos, G.D.; et al. VEGF-mediated inflammation precedes angiogenesis in adult brain. Exp. Neurol. 2004, 187, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Marumo, T.; Schini-Kerth, V.B.; Busse, R. Vascular endothelial growth factor activates nuclear factor-kappab and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 1999, 48, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Avraham, H.; Lee, S.H.; Avraham, S. Vascular endothelial growth factor modulates neutrophil transendothelial migration via up-regulation of interleukin-8 in human brain microvascular endothelial cells. J. Biol. Chem. 2002, 277, 10445–10451. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Zhang, X.; Li, Y.; Xu, H. Toll-like receptor 3 activation drives the inflammatory response in oxygen-induced retinopathy in rats. Br. J. Ophthalmol. 2015, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Storkebaum, E.; de Almodovar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Singh, R.S.; Fort, P.E.; Gardner, T.W. Neuroprotection for diabetic retinopathy. Dev. Ophthalmol. 2009, 44, 56–68. [Google Scholar] [PubMed]
- Gilbert, R.E.; Vranes, D.; Berka, J.L.; Kelly, D.J.; Cox, A.; Wu, L.L.; Stacker, S.A.; Cooper, M.E. Vascular endothelial growth factor and its receptors in control and diabetic rat eyes. Lab. Investig. 1998, 78, 1017–1027. [Google Scholar] [PubMed]
- Barber, A.J.; Nakamura, M.; Wolpert, E.B.; Reiter, C.E.; Seigel, G.M.; Antonetti, D.A.; Gardner, T.W. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J. Biol. Chem. 2001, 276, 32814–32821. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.C.; Bazan, N.G. Mediator lipidomics in ophthalmology: Targets for modulation in inflammation, neuroprotection and nerve regeneration. Curr. Eye Res. 2013, 38, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Tanaka, T.; Nawa, H.; Usui, T.; Fukuchi, T.; Ikeda, K.; Abe, H.; Takei, N. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: Therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes 2004, 53, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Deng, Q.Q.; Wu, X.H.; Yu, J.; Yang, X.L.; Zhong, Y.M. Upregulation of glutamate-aspartate transporter by glial cell line-derived neurotrophic factor ameliorates cell apoptosis in neural retina in streptozotocin-induced diabetic rats. CNS Neurosci. Ther. 2013, 19, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Aizu, Y.; Katayama, H.; Takahama, S.; Hu, J.; Nakagawa, H.; Oyanagi, K. Topical instillation of ciliary neurotrophic factor inhibits retinal degeneration in streptozotocin-induced diabetic rats. Neuroreport 2003, 14, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.K.; Al-Gayyar, M.M.; Matragoon, S.; Pillai, B.A.; Abdelsaid, M.A.; Nussbaum, J.J.; El-Remessy, A.B. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia 2011, 54, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.N.; Vindeirinho, J.; Cavadas, C.; Ambrosio, A.F.; Santos, P.F. Contribution of tnf receptor 1 to retinal neural cell death induced by elevated glucose. Mol. Cell. Neurosci. 2012, 50, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-induced pro-inflammatory responses by retinal muller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Howell, S.J.; Hatala, D.A.; Huang, K.; Kern, T.S. Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes 2007, 56, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Berkowitz, B.A. Photoreceptors in diabetic retinopathy. J. Diabetes Investig. 2015, 6, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Tonade, D.; Liu, H.; Kern, T.S. Photoreceptor cells produce inflammatory mediators that contribute to endothelial cell death in diabetes. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4264–4271. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Sun, H.L.; Wu, L.M.; Guo, X.J.; Dou, H.L.; Tso, M.O.; Zhao, L.; Li, S.M. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.U.; Jackson, G.R.; Quillen, D.A.; Larsen, M.; Klein, R.; Liao, J.; Holfort, S.; Munch, I.C.; Gardner, T.W. Effect of doxycycline vs placebo on retinal function and diabetic retinopathy progression in patients with severe nonproliferative or non-high-risk proliferative diabetic retinopathy: A randomized clinical trial. JAMA Ophthalmol. 2014, 132, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. Cd40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef] [PubMed]
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Portillo, J.C.; Lopez Corcino, Y.; Miao, Y.; Tang, J.; Sheibani, N.; Kern, T.S.; Dubyak, G.R.; Subauste, C.S. CD40 in retinal muller cells induces P2X7-dependent cytokine expression in macrophages/microglia in diabetic mice and development of early experimental diabetic retinopathy. Diabetes 2017, 66, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Samuels, I.S.; Portillo, J.C.; Miao, Y.; Kern, T.S.; Subauste, C.S. Loss of CD40 attenuates experimental diabetes-induced retinal inflammation but does not protect mice from electroretinogram defects. Vis. Neurosci. 2017, 34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tang, J.; Du, Y.; Lee, C.A.; Kern, T.S. Beneficial effects of a novel rage inhibitor on early diabetic retinopathy and tactile allodynia. Mol. Vis. 2011, 17, 3156–3165. [Google Scholar] [PubMed]
- Lee, C.A.; Li, G.; Patel, M.D.; Petrash, J.M.; Benetz, B.A.; Veenstra, A.; Amengual, J.; von Lintig, J.; Burant, C.J.; Tang, J.; et al. Diabetes-induced impairment in visual function in mice: Contributions of p38 mapk, rage, leukocytes, and aldose reductase. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2904–2910. [Google Scholar] [CrossRef] [PubMed]
- Calderon, G.D.; Juarez, O.H.; Hernandez, G.E.; Punzo, S.M.; De la Cruz, Z.D. Oxidative stress and diabetic retinopathy: Development and treatment. Eye (London) 2017, 31, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, F.S.; Allkabes, M.; Salsini, G.; Bonifazzi, C.; Perri, P. The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci. 2016, 162, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.Y.; Green, W.R.; Tso, M.O. Microglial activation in human diabetic retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.H.; Draeby, D.; Owens, T. Microglia are required for astroglial toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia 2012, 60, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, N.; Fernandez-Sanchez, L.; Campello, L.; Maneu, V.; De la Villa, P.; Lax, P.; Pinilla, I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog. Retin. Eye Res. 2014, 43, 17–75. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Lelios, I.; Croxford, A.L. Microglia versus myeloid cell nomenclature during brain inflammation. Front. Immunol. 2015, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Miller-Fleming, L.; Pais, T.F. Microglia and inflammation: Conspiracy, controversy or control? Cell. Mol. Life Sci. 2014, 71, 3969–3985. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Foster, T.D.; Thomas, W.E. Cellular forms and functions of brain microglia. Brain Res. Bull. 1994, 34, 73–78. [Google Scholar] [CrossRef]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of microglia in cns inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; El-Remessy, A.B.; Matragoon, S.; Zhang, W.; Patel, Y.; Khan, S.; Al-Gayyar, M.M.; El-Shishtawy, M.M.; Liou, G.I. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes 2011, 60, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.; Allen, D.M.; Cardona, A.E. The role of microglia in diabetic retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef] [PubMed]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Coorey, N.J.; Shen, W.; Chung, S.H.; Zhu, L.; Gillies, M.C. The role of glia in retinal vascular disease. Clin. Exp. Optom. 2012, 95, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.; Reichenbach, A. The muller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Eastlake, K.; Banerjee, P.J.; Angbohang, A.; Charteris, D.G.; Khaw, P.T.; Limb, G.A. Muller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 2016, 64, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Gerhardinger, C.; Costa, M.B.; Coulombe, M.C.; Toth, I.; Hoehn, T.; Grosu, P. Expression of acute-phase response proteins in retinal muller cells in diabetes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, F.; Xiong, H.; Hu, D.; Limb, G.A.; Xie, T.; Peng, L.; Yang, W.; Sun, Y.; Zhou, M.; et al. Il-1beta upregulates IL-8 production in human muller cells through activation of the p38 mapk and erk1/2 signaling pathways. Inflammation 2014, 37, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Ridet, J.L.; Malhotra, S.K.; Privat, A.; Gage, F.H. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997, 20, 570–577. [Google Scholar] [CrossRef]
- Rungger-Brandle, E.; Messerli, J.M.; Niemeyer, G.; Eppenberger, H.M. Confocal microscopy and computer-assisted image reconstruction of astrocytes in the mammalian retina. Eur. J. Neurosci. 1993, 5, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, J.H.; Park, J.A.; Lee, S.W.; Kim, W.J.; Yu, Y.S.; Kim, K.W. Blood-neural barrier: Intercellular communication at glio-vascular interface. J. Biochem. Mol. Biol. 2006, 39, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.S.; Huang, Q.; Gurel, Z.; Sorenson, C.M.; Sheibani, N. High glucose alters retinal astrocytes phenotype through increased production of inflammatory cytokines and oxidative stress. PLoS ONE 2014, 9, e103148. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. Control of autoimmune cns inflammation by astrocytes. Semin. Immunopathol. 2015, 37, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; LaNoue, K.F.; Berkich, D.A.; Xu, B.; Ratz, M.; Taylor, C.; Hutson, S.M. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 2001, 76, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Orkand, R.K.; Nicholls, J.G.; Kuffler, S.W. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 788–806. [Google Scholar] [CrossRef] [PubMed]
- Uckermann, O.; Wolf, A.; Kutzera, F.; Kalisch, F.; Beck-Sickinger, A.G.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Glutamate release by neurons evokes a purinergic inhibitory mechanism of osmotic glial cell swelling in the rat retina: Activation by neuropeptide y. J. Neurosci. Res. 2006, 83, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Metea, M.R.; Newman, E.A. Glial cells dilate and constrict blood vessels: A mechanism of neurovascular coupling. J. Neurosci. 2006, 26, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L.; Wu, C.H.; Jiang-Shieh, Y.F.; Shieh, J.Y.; Wen, C.Y. Reactive changes of retinal astrocytes and muller glial cells in kainate-induced neuroexcitotoxicity. J. Anat. 2007, 210, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, P.M.; Stevens, B. Microglia function during brain development: New insights from animal models. Brain Res. 2015, 1617, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.J.; Le, Y.Z. Muller glia are a major cellular source of survival signals for retinal neurons in diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in muller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Amin, S.; Roy, S. Retinal fibrosis in diabetic retinopathy. Exp. Eye Res. 2016, 142, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Fogal, B.; Hewett, S.J. Interleukin-1beta: A bridge between inflammation and excitotoxicity? J. Neurochem. 2008, 106, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Tnf-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [PubMed]
- Abcouwer, S.F.; Shanmugam, S.; Gomez, P.F.; Shushanov, S.; Barber, A.J.; Lanoue, K.F.; Quinn, P.G.; Kester, M.; Gardner, T.W. Effect of IL-1beta on survival and energy metabolism of R28 and RGC-5 retinal neurons. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5581–5592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sheridan, G.K.; Murphy, K.J. Neuron-glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Sun, J.H.; Xiang, F.F.; Liu, L.; Li, W.J. Aquaporin 4 knockdown exacerbates streptozotocin-induced diabetic retinopathy through aggravating inflammatory response. Exp. Eye Res. 2012, 98, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Tolentino, M.J.; McLeod, D.S.; Taomoto, M.; Otsuji, T.; Adamis, A.P.; Lutty, G.A. Pathologic features of vascular endothelial growth factor-induced retinopathy in the nonhuman primate. Am. J. Ophthalmol. 2002, 133, 373–385. [Google Scholar] [CrossRef]
- Simo, R.; Hernandez, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Retin. Eye Res. 2015, 48, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Unterlauft, J.D.; Claudepierre, T.; Schmidt, M.; Muller, K.; Yafai, Y.; Wiedemann, P.; Reichenbach, A.; Eichler, W. Enhanced survival of retinal ganglion cells is mediated by muller glial cell-derived PEDF. Exp. Eye Res. 2014, 127, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Unterlauft, J.D.; Eichler, W.; Kuhne, K.; Yang, X.M.; Yafai, Y.; Wiedemann, P.; Reichenbach, A.; Claudepierre, T. Pigment epithelium-derived factor released by muller glial cells exerts neuroprotective effects on retinal ganglion cells. Neurochem. Res. 2012, 37, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Zhang, X.M.; Liu, J.J.; Dong, L.; Feng, Z.L. Effect of high glucose concentration on VEGF and PEDF expression in cultured retinal muller cells. Mol. Biol. Rep. 2009, 36, 2147–2151. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.; Yafai, Y.; Reichenbach, A.; Wiedemann, P.; Eichler, W. Regulation of pigment epithelium-derived factor production and release by retinal glial (muller) cells under hypoxia. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5161–5167. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Miyagi, M.; Darrow, R.M.; Crabb, J.S.; Hollyfield, J.G.; Organisciak, D.T.; Crabb, J.W. Intense light exposure changes the crystallin content in retina. Exp. Eye Res. 2003, 76, 131–133. [Google Scholar] [CrossRef]
- Sax, C.M.; Piatigorsky, J. Expression of the alpha-crystallin/small heat-shock protein/molecular chaperone genes in the lens and other tissues. Adv. Enzymol. Relat. Areas Mol. Biol. 1994, 69, 155–201. [Google Scholar] [PubMed]
- Rao, N.A.; Saraswathy, S.; Wu, G.S.; Katselis, G.S.; Wawrousek, E.F.; Bhat, S. Elevated retina-specific expression of the small heat shock protein, alphaa-crystallin, is associated with photoreceptor protection in experimental uveitis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.R.; Inman, D.M.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Chona, F.; Song, B.K.; Geisert, E.E., Jr. Temporal changes in gene expression after injury in the rat retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Ruebsam, A.; Dulle, J.E.; Myers, A.M.; Sakrikar, D.; Green, K.M.; Khan, N.W.; Schey, K.; Fort, P.E. A specific phosphorylation regulates the protective role of alphaa-crystallin in diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Landon, M.; Pike, I.; Spendlove, I.; McDermott, H.; Mayer, R.J. Dementia with beta-amyloid deposition: Involvement of alpha b-crystallin supports two main diseases. Lancet 1990, 336, 515–516. [Google Scholar] [CrossRef]
- Renkawek, K.; Voorter, C.E.; Bosman, G.J.; van Workum, F.P.; de Jong, W.W. Expression of alpha b-crystallin in Alzheimer’s disease. Acta Neuropathol. 1994, 87, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Wisniewski, T.; Iwaki, A.; Corbin, E.; Tomokane, N.; Tateishi, J.; Goldman, J.E. Accumulation of alpha b-crystallin in central nervous system glia and neurons in pathologic conditions. Am. J. Pathol. 1992, 140, 345–356. [Google Scholar] [PubMed]
- Kegel, K.B.; Iwaki, A.; Iwaki, T.; Goldman, J.E. Alphab-crystallin protects glial cells from hypertonic stress. Am. J. Physiol. 1996, 270, C903–C909. [Google Scholar] [CrossRef] [PubMed]
- Parks, W.C.; Wilson, C.L.; Lopez-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Kashiwagi, K.; Iizuka, Y.; Tanaka, Y.; Imai, M.; Tsukahara, S. Matrix metalloproteinases in human diabetic and nondiabetic vitreous. Retina 2001, 21, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Liu, H.; Williams, I.; Chaqour, B. Matrix metalloproteinase-2 expression and apoptogenic activity in retinal pericytes: Implications in diabetic retinopathy. Ann. N. Y. Acad. Sci. 2007, 1103, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Trivedi, A.; Lee, J.U.; Lohela, M.; Lee, S.M.; Fandel, T.M.; Werb, Z.; Noble-Haeusslein, L.J. Matrix metalloproteinase-9 and stromal cell-derived factor-1 act synergistically to support migration of blood-borne monocytes into the injured spinal cord. J. Neurosci. 2011, 31, 15894–15903. [Google Scholar] [CrossRef] [PubMed]
- Navaratna, D.; McGuire, P.G.; Menicucci, G.; Das, A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes 2007, 56, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Z.; Xue, Z.; Zhu, Y.; Yang, G.Y.; Young, W.L. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke 2007, 38, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Sears, D.D. TLR4 and insulin resistance. Gastroenterol. Res. Pract. 2010, 2010, 212563. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (rage) signaling pathways via high mobility group b1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Ju, M.; Lee, K.Y.V.; Mackey, A.; Evangelista, M.; Iwata, D.; Adamson, P.; Lashkari, K.; Foxton, R.; Shima, D.; et al. A proinflammatory function of toll-like receptor 2 in the retinal pigment epithelium as a novel target for reducing choroidal neovascularization in age-related macular degeneration. Am. J. Pathol. 2017, 187, 2208–2221. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, U.; Jialal, I. Hyperglycemia induces toll-like receptor-2 and -4 expression and activity in human microvascular retinal endothelial cells: Implications for diabetic retinopathy. J. Diabetes Res. 2014, 2014, 790902. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.B.; Chen, X.; Timothy, N.; Aiello, L.P.; Feener, E.P. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J. Proteome Res. 2008, 7, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ramirez, M.; Canals, F.; Hernandez, C.; Colome, N.; Ferrer, C.; Carrasco, E.; Garcia-Arumi, J.; Simo, R. Proteomic analysis of human vitreous fluid by fluorescence-based difference gel electrophoresis (DIGE): A new strategy for identifying potential candidates in the pathogenesis of proliferative diabetic retinopathy. Diabetologia 2007, 50, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Bu, H.; Portillo, J.A.; Li, Y.; Subauste, C.S.; Huang, S.S.; Kern, T.S.; Lin, F. Modulation of retinal muller cells by complement receptor c5ar. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8191–8198. [Google Scholar] [CrossRef] [PubMed]
- King, P.; Peacock, I.; Donnelly, R. The uk prospective diabetes study (UKPDS): Clinical and therapeutic implications for type 2 diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648. [Google Scholar] [CrossRef] [PubMed]
- The effect of intensive diabetes treatment on the progression of diabetic retinopathy in insulin-dependent diabetes mellitus. The diabetes control and complications trial. Arch. Ophthalmol. 1995, 113, 36–51. [Google Scholar]
- Cunningham, M.A.; Edelman, J.L.; Kaushal, S. Intravitreal steroids for macular edema: The past, the present, and the future. Surv. Ophthalmol. 2008, 53, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B. Intravitreal triamcinolone acetonide for diabetic retinopathy. Dev. Ophthalmol. 2007, 39, 96–110. [Google Scholar] [PubMed]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Kirchhof, B.; Koizumi, K.; Dohmen, S.; Adamis, A.P. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002, 16, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Sarthy, V.P.; Kern, T.S. Interaction between NO and COX pathways in retinal cells exposed to elevated glucose and retina of diabetic rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R735–R741. [Google Scholar] [CrossRef] [PubMed]
- Early Treatment Diabetic Retinopathy Study Research Group. Effects of aspirin treatment on diabetic retinopathy. Etdrs report number 8. Ophthalmology 1991, 98, 757–765. [Google Scholar]
- The Damad Study Group. Effect of aspirin alone and aspirin plus dipyridamole in early diabetic retinopathy. A multicenter randomized controlled clinical trial. Diabetes 1989, 38, 491–498. [Google Scholar]
- Kim, S.J.; Flach, A.J.; Jampol, L.M. Nonsteroidal anti-inflammatory drugs in ophthalmology. Surv. Ophthalmol. 2010, 55, 108–133. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Miller, C.M.; Du, Y.; Zheng, L.; Mohr, S.; Ball, S.L.; Kim, M.; Jamison, J.A.; Bingaman, D.P. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes 2007, 56, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Amrite, A.C.; Ayalasomayajula, S.P.; Cheruvu, N.P.; Kompella, U.B. Single periocular injection of celecoxib-plga microparticles inhibits diabetes-induced elevations in retinal PGE2, VEGF, and vascular leakage. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Tsilimbaris, M.K.; Panagiotoglou, T.D.; Charisis, S.K.; Anastasakis, A.; Krikonis, T.S.; Christodoulakis, E. The use of intravitreal etanercept in diabetic macular oedema. Semin. Ophthalmol. 2007, 22, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Markomichelakis, N.; Theodossiadis, G.P.; Grigoropoulos, V.; Katsilambros, N.; Theodossiadis, P.G. Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care 2005, 28, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Stahel, M.; Becker, M.; Graf, N.; Michels, S. Systemic interleukin 1beta inhibition in proliferative diabetic retinopathy: A prospective open-label study using canakinumab. Retina 2016, 36, 385–391. [Google Scholar] [CrossRef] [PubMed]
- A Phase 2, Multi-Center Study to Compare the Efficacy and Safety of a Chemokine CCR2/5 Receptor Antagonist with Ranibizumab in Adults Withdiabetic Macular Edema. Available online: https://clinicaltrials.gov/ct2/show/NCT01994291 (accessed on 30 October 2017).
- Ulbrich, H.; Eriksson, E.E.; Lindbom, L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol. Sci. 2003, 24, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Allegro Ophthalmics Announces Positive Topline Results from Del Mar Phase 2b Trial Evaluating Luminate® in Patients with Diabetic Macular Edema. Available online: http://www.allegroeye.com/press (accessed on 30 October 2017).
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 sensitizes endothelial cells to tnf-alpha and has a crucial role in the induction of inflammation. Nat. Med. 2006, 12, 235–239. [Google Scholar] [CrossRef] [PubMed]
- The Time-2b Study: A Study of Akb-9778, a Novel Tie 2 Activator, in Patients with Non-Proliferative Diabetic Retinopathy (NPDR) (Time-2b). Available online: https://clinicaltrials.gov/ct2/show/NCT03197870 (accessed on 30 October 2017).
- Sjolie, A.K.; Klein, R.; Porta, M.; Orchard, T.; Fuller, J.; Parving, H.H.; Bilous, R.; Chaturvedi, N.; DIRECT Programme Study Group. Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (direct-protect 2): A randomised placebo-controlled trial. Lancet 2008, 372, 1385–1393. [Google Scholar] [CrossRef]
- Mauer, M.; Zinman, B.; Gardiner, R.; Suissa, S.; Sinaiko, A.; Strand, T.; Drummond, K.; Donnelly, S.; Goodyer, P.; Gubler, M.C.; et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N. Engl. J. Med. 2009, 361, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Mesa, N.; Zarzuelo, A.; Galvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Matute, C. Neuroprotection by tetracyclines. Trends Pharmacol. Sci. 2004, 25, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, A.L.; Kaushal, D.; Philipp, M.T. The antibiotics doxycycline and minocycline inhibit the inflammatory responses to the lyme disease spirochete borrelia burgdorferi. J. Infect. Dis. 2009, 199, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Cukras, C.A.; Petrou, P.; Chew, E.Y.; Meyerle, C.B.; Wong, W.T. Oral minocycline for the treatment of diabetic macular edema (DME): Results of a phase I/II clinical study. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Lachin, J.M.; Genuth, S.; Nathan, D.M.; Zinman, B.; Rutledge, B.N.; Group, D.E.R. Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial—Revisited. Diabetes 2008, 57, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Du, Y.; Lee, C.A.; Talahalli, R.; Eells, J.T.; Kern, T.S. Low-intensity far-red light inhibits early lesions that contribute to diabetic retinopathy: In vivo and in vitro. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3681–3690. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Herda, A.A.; Kern, T.S. Photobiomodulation in the treatment of patients with non-center-involving diabetic macular oedema. Br. J. Ophthalmol. 2014, 98, 1013–1015. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Diabetic Retinopathy Stage | References | |||
|---|---|---|---|---|---|
| Diabetics without DR | NPDR | PDR | DME | ||
| AQP4 | ↑ | ↑ | not known | not known | [47] |
| GFAP | ↑ | ↑ | not known | not known | [47] |
| VEGF | ↑ serum ↔ vitreous | ↑ | ↑ | ↑ | [68,69,70] |
| IFN-γ | ↑ | ↑ | ↑ | ↑ | [49,52,71,72] |
| IL-1α | ↑ | ↑ | ↑ | ↑ | [49,69] |
| IL-3 | ↑ vitreous ↓ serum | ↓ vitreous & serum | ↓ serum | ↑ aqueous no change vitreous | [49,73,74,75] |
| s-IL2R | ↑ | ↑ | ↑ | not known | [56,73] |
| MCP-2 | ↑ | ↑ | not known | not known | [49] |
| ICAM-1 | ↑ serum | ↑ serum | ↑ | ↑ | [68,70,71] |
| IL-1β | no | ↑ | ↑ | ↑ | [52,76] |
| IL-6 | no | ↑ | ↑ | ↑ | [52,69,70,77] |
| IL-8 | no | ↑↑ | ↑ | ↑ | [51,69,77,78] |
| IL-2 | no | ↑ | ↑ | not known | [52,79] |
| IL-4 | no | ↑ | ↑ | not known | [52,80] |
| IL-5 | no | ↑ | ↑ | not known | [52] |
| MCP-1 | no | ↑ | ↑ | ↑ | [49,77] |
| TNF-α | no | ↑↑ | ↑ | ↑ | [51,52,70] |
| sTNF-R | no | ↑ | ↑ | not known | [49] |
| RANTES | no | ↑ | ↑ | ↑ | [49,81] |
| IP-10 | no | ↑ | ↑ | ↑ | [49,77,82] |
| GM-CSF | no | ↑ | ↑ | not known | [49,69] |
| PEDF | ↔ vitreous | ↔ serum | ↑active PDR ↓ inactive PDR | ↓ vitreous | [12,83,84,85,86] |
| IGF-1 | no | conflicting results | ↑ | not known | [87,88,89] |
| PlGF | no | no | ↑ | ↑ | [90] |
| IL-10 | no | no | ↑ | ↑ | [52,69,76] |
| Complement factors | no | no | ↑ | ↑ | [17,63] |
| bFGF | not known | not known | conflicting results | conflicting results | [89,91] |
| CD40 | no | ↑ | ↑ | not known | [92] |
| HIF-1α | not known | ↑ | ↑ | not known | [93,94] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. https://doi.org/10.3390/ijms19040942
Rübsam A, Parikh S, Fort PE. Role of Inflammation in Diabetic Retinopathy. International Journal of Molecular Sciences. 2018; 19(4):942. https://doi.org/10.3390/ijms19040942
Chicago/Turabian StyleRübsam, Anne, Sonia Parikh, and Patrice E. Fort. 2018. "Role of Inflammation in Diabetic Retinopathy" International Journal of Molecular Sciences 19, no. 4: 942. https://doi.org/10.3390/ijms19040942
APA StyleRübsam, A., Parikh, S., & Fort, P. E. (2018). Role of Inflammation in Diabetic Retinopathy. International Journal of Molecular Sciences, 19(4), 942. https://doi.org/10.3390/ijms19040942