Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms


{kind=link}
{kind=link}
Abstract
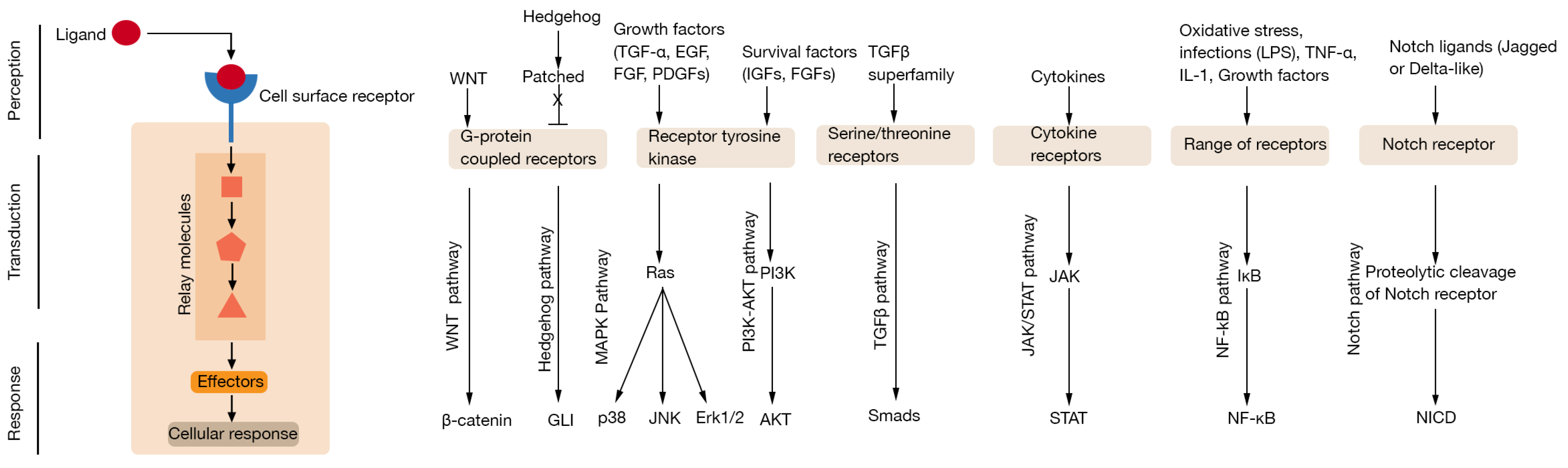
1. Introduction
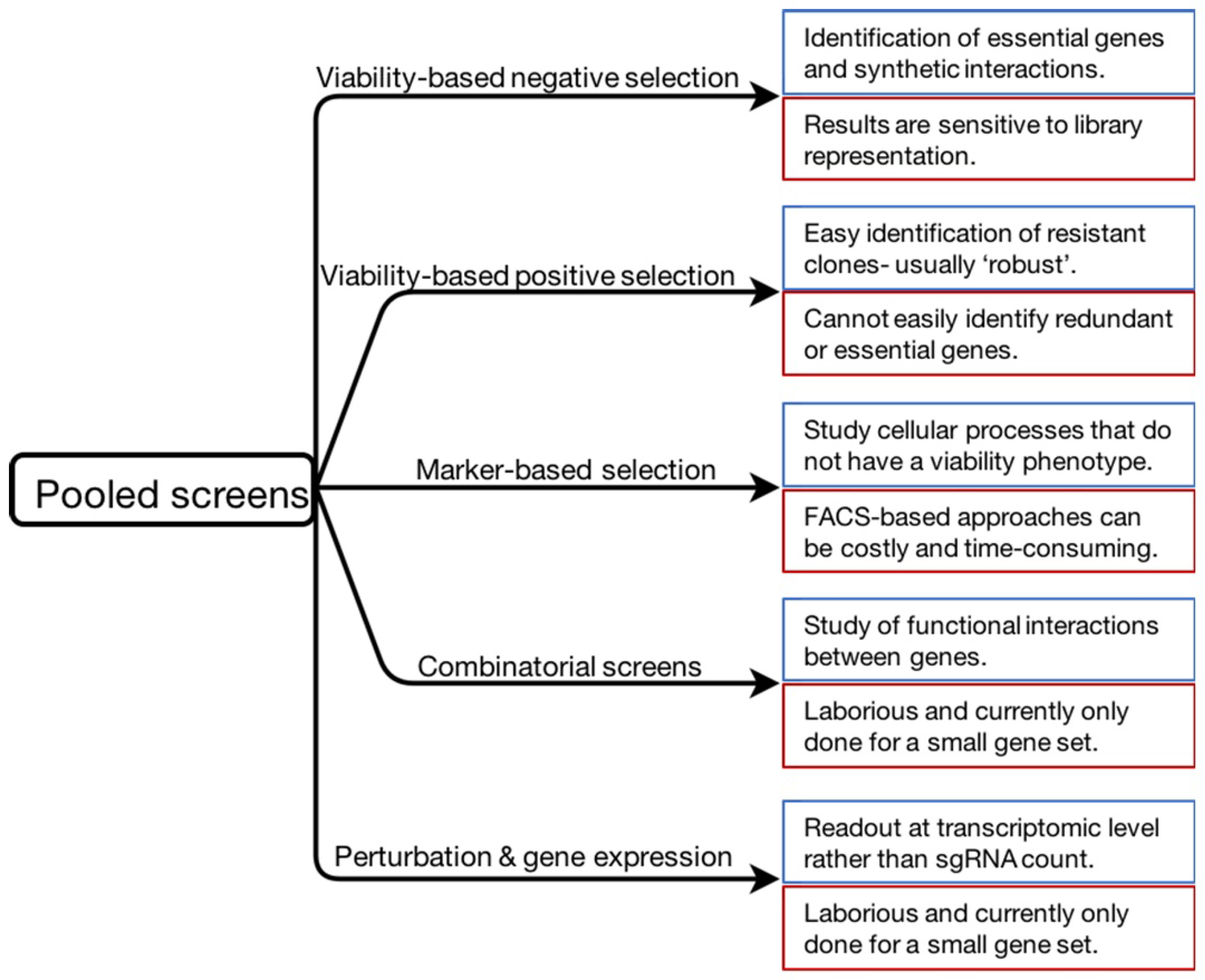
2. Different Strands of Pooled CRISPR Screens
2.1. Viability-Based Negative-Selection Screens
2.2. Viability-Based Positive-Selection Screens
2.3. Marker-Based Selection Screen
2.4. Combinatorial Screens
3. Conclusions and Further Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| BMDC | Bone marrow-derived dendritic cells |
| cAMP | Cyclic adenosine monophosphate |
| CRISPR/Cas9 | Clustered regularly-interspaced short palindromic repeats/Cas9 |
| Erk | Extracellular signal-regulated protein or externally-regulated kinases |
| EGFR | Epidermal growth factor receptor |
| FACS | Fluorescence-activated cell sorting |
| FGF | Fibroblast growth factor |
| GFP | Green fluorescent protein |
| GPCR | G-protein–coupled receptors |
| JNK | Jun N-terminal kinase |
| JAK | Janus Kinase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| NICD | Notch intracellular domain |
| NFB | Nuclear factor B |
| OST | Oligosaccharyltransferase |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphatidylinositol 3 kinase |
| RNAi | RNA interference |
| RTK | Receptor tyrosine kinase |
| SAM | Synergistic activation mediator |
| SHH | Sonic Hedgehog |
| Smads | Sma and mad homology signal proteins |
| TGF | Transforming growth factor beta |
| TNF | Tumour necrosis factor |
References
- Hunter, T. Signaling-2000 and beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef]
- Finkel, T.; Silvio Gutkind, J. Signal Transduction and Human Disease; John Wiley & Sons: New York, NY, USA, 2003. [Google Scholar]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Shay, T.; O’Shea, E.K.; Regev, A. Transcriptional regulatory circuits: Predicting numbers from alphabets. Science 2009, 325, 429–432. [Google Scholar] [PubMed]
- Amit, I.; Regev, A.; Hacohen, N. Strategies to discover regulatory circuits of the mammalian immune system. Nat. Rev. Immunol. 2011, 11, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Bandyopadhyay, S.; Ideker, T. Integrating physical and genetic maps: from genomes to interaction networks. Nat. Rev. Genet. 2007, 8, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Mann, M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 2010, 11, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Spurrier, B.; Ramalingam, S.; Nishizuka, S. Reverse-phase protein lysate microarrays for cell signalling analysis. Nat. Protoc. 2008, 3, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Module 2: Cell Signalling Pathways. Cell Signal. Biol. 2014, 6, csb0001002. [Google Scholar] [CrossRef]
- Doench, J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018, 19, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knock-out and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knock-out screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Moffat, J. BAGEL: A computational framework for identifying essential genes from pooled library screens. BMC Bioinform. 2016, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Breinig, M.; Heigwer, F.; Brügemann, D.; Leible, S.; Pelz, O.; Zhan, T.; Boutros, M. caRpools: An R package for exploratory data analysis and documentation of pooled CRISPR/Cas9 screens. Bioinformatics 2016, 32, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Qi, L. Genetic and epigenetic control of gene expression by CRISPR-Cas systems. F1000Research 2017, 6, 747. [Google Scholar]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Alexe, G.; Dharia, N.V.; Ross, L.; Iniguez, A.B.; Conway, A.S.; Wang, E.J.; Veschi, V.; Lam, N.; Qi, J.; et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Investig. 2018, 128, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Deregulation of cell signalling in cancer. FEBS Lett. 2014, 588, 2558–2570. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt–FZD5 signalling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumours. Nat. Med. 2016, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.M.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR knock-out screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Munoz, D.M.; Cassiani, P.J.; Li, L.; Billy, E.; Korn, J.M.; Jones, M.D.; Golji, J.; Ruddy, D.A.; Yu, K.; McAllister, G.; et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016, 6, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B. Genomic copy number dictates a gene-independent cell response to CRISPR-Cas9 targeting. Cancer Discov. 2016, 6, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knock-out screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Wu, C.; Wang, Y.; Qi, R.; Bhavanasi, D.; Zuo, Z.; Dos Santos, C.; Chen, S.; Chen, Y.; Zheng, H.; et al. A Genome-Wide CRISPR Screen Identifies Genes Critical for Resistance to FLT3 Inhibitor AC220. Cancer Res. 2017, 77, 4402–4413. [Google Scholar] [CrossRef] [PubMed]
- Koike-Yusa, H.; Li, Y.; Tan, E.P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.C.J.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Haga, K.; Fujimoto, A.; Takai-Todaka, R.; Miki, M.; Doan, Y.H.; Murakami, K.; Yokoyama, M.; Murata, K.; Nakanishi, A.; Katayama, K. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6248–E6255. [Google Scholar] [CrossRef] [PubMed]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife 2017, 6, e18970. [Google Scholar] [PubMed]
- Terai, H.; Kitajima, S.; Potter, D.S.; Matsui, Y.; Gutierrez Quiceno, L.; Chen, T.; Kim, T.J.; Rusan, M.; Thai, T.C.; Piccioni, F.; et al. ER stress signalling promotes the survival of cancer ‘persister cells’ tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. 2017, 78, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Krall, E.B.; Aguirre, A.J.; Kim, M.; Widlund, H.R.; Doshi, M.B.; Sicinska, E.; Sulahian, R.; Goodale, A.; Cowley, G.S.; et al. ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep. 2017, 18, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.Y.; Niessen, H.W.M.; Rozeman, E.A.; Geukes Foppen, M.H.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Hoogendoorn, S.; Kopp, A.R.; Morgens, D.W.; Vu, B.K.; Kennedy, M.C.; Han, K.; Li, A.; Hess, G.T.; Bassik, M.C.; et al. A CRISPR-based screen for Hedgehog signalling provides insights into ciliary function and ciliopathies. Nat. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Parnas, O.; Jovanovic, M.; Eisenhaure, T.M.; Herbst, R.H.; Dixit, A.; Ye, C.J.; Przybylski, D.; Platt, R.J.; Tirosh, I.; Sanjana, N.E.; et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 2015, 162, 675–686. [Google Scholar] [CrossRef] [PubMed]
- DeJesus, R.; Moretti, F.; McAllister, G.; Wang, Z.; Bergman, P.; Liu, S.; Frias, E.; Alford, J.; Reece-Hoyes, J.S.; Lindeman, A.; et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Potting, C.; Crochemore, C.; Moretti, F.; Nigsch, F.; Schmidt, I.; Manneville, C.; Carbone, W.; Knehr, J.; DeJesus, R.; Lindeman, A.; et al. Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E180–E189. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 2018, 44, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Lebensohn, A.M.; Dubey, R.; Neitzel, L.R.; Tacchelly-Benites, O.; Yang, E.; Marceau, C.D.; Davis, E.M.; Patel, B.B.; Bahrami-Nejad, Z.; Travaglini, K.J.; et al. Comparative genetic screens in human cells reveal new regulatory mechanisms in WNT signalling. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Billmann, M.; Chaudhary, V.; ElMaghraby, M.F.; Fischer, B.; Boutros, M. Widespread Rewiring of Genetic Networks upon Cancer Signaling Pathway Activation. Cell Syst. 2018, 6, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Horn, T.; Sandmann, T.; Fischer, B.; Axelsson, E.; Huber, W.; Boutros, M. Mapping of signalling networks through synthetic genetic interaction analysis by RNAi. Nat. Methods 2011, 8, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.L.; Choi, G.C.G.; Cui, C.H.; Pregernig, G.; Milani, P.; Adam, M.; Perli, S.D.; Kazer, S.W.; Gaillard, A.; Hermann, M.; et al. Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc. Natl. Acad. Sci. USA 2016, 113, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.L.; Choi, G.C.G.; Cheng, A.A.; Purcell, O.; Lu, T.K. Massively parallel high-order combinatorial genetics in human cells. Nat. Biotechnol. 2015, 33, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017, 35, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.P.; Zhao, D.; Sasik, R.; Luebeck, J.; Birmingham, A.; Bojorquez-Gomez, A.; Licon, K.; Klepper, K.; Pekin, D.; Beckett, A.N.; et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 2017, 14, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Roguev, A.; Gordon, D.E.; Chen, M.; Chen, S.H.; Shales, M.; Shen, J.P.; Ideker, T.; Mali, P.; Qi, L.S.; et al. Genetic interaction mapping in mammalian cells using CRISPR interference. Nat. Methods 2017, 14, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, J.E.; Abudayyeh, O.O.; Joung, J.; Gootenberg, J.S.; Zhang, F.; Konermann, S. Orthogonal gene knock-out and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015, 33, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; Tian, R.; Blau, J.A.; Markegard, E.; Wagner, R.T.; Wu, D.; Mo, X.; Biton, A.; Zaitlen, N.; Fu, H.; et al. Dual gene activation and knock-out screen reveals directional dependencies in genetic networks. Nat. Biotechnol. 2018, 36, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Unniyampurath, U.; Pilankatta, R.; Krishnan, M.N. RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi? Int. J. Mol. Sci. 2016, 17, 291. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Bartholdson, S.J.; Couch, A.C.; Yusa, K.; Wright, G.J. Genome-Scale Identification of Cellular Pathways Required for Cell Surface Recognition. Submitted.
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Q.; Li, Y.; Mou, H.; Moore, J.; Park, A.; Pomyen, Y.; Hough, S.; Kennedy, Z.; Fischer, A.; Yin, H.; et al. Genome-Wide CRISPR Screen Identifies Regulators of Mitogen-Activated Protein Kinase as Suppressors of Liver Tumors in Mice. Gastroenterology 2017, 152, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329. [Google Scholar] [CrossRef] [PubMed]
- Yau, E.H.; Kummetha, I.R.; Lichinchi, G.; Tang, R.; Zhang, Y.; Rana, T.M. Genome-Wide CRISPR Screen for Essential Cell Growth Mediators in Mutant KRAS Colorectal Cancers. Cancer Res. 2017, 77, 6330–6339. [Google Scholar] [CrossRef] [PubMed]
- Michlits, G.; Hubmann, M.; Wu, S.H.; Vainorius, G.; Budusan, E.; Zhuk, S.; Burkard, T.R.; Novatchkova, M.; Aichinger, M.; Lu, Y.; et al. CRISPR-UMI: single-cell lineage tracing of pooled CRISPR-Cas9 screens. Nat. Methods 2017, 14, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167, 1867–1882. [Google Scholar] [CrossRef] [PubMed]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016, 167, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Duan, J.; Li, B.; Zhou, P.; Hon, G.C. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 2017, 66, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Perli, S.D.; Cui, C.H.; Lu, T.K. Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. Int. J. Mol. Sci. 2018, 19, 933. https://doi.org/10.3390/ijms19040933
Sharma S, Petsalaki E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 2018; 19(4):933. https://doi.org/10.3390/ijms19040933
Chicago/Turabian StyleSharma, Sumana, and Evangelia Petsalaki. 2018. "Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms" International Journal of Molecular Sciences 19, no. 4: 933. https://doi.org/10.3390/ijms19040933
APA StyleSharma, S., & Petsalaki, E. (2018). Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences, 19(4), 933. https://doi.org/10.3390/ijms19040933