iAPSL-IF: Identification of Apoptosis Protein Subcellular Location Using Integrative Features Captured from Amino Acid Sequences

Abstract
:1. Introduction
2. Results and Discussion
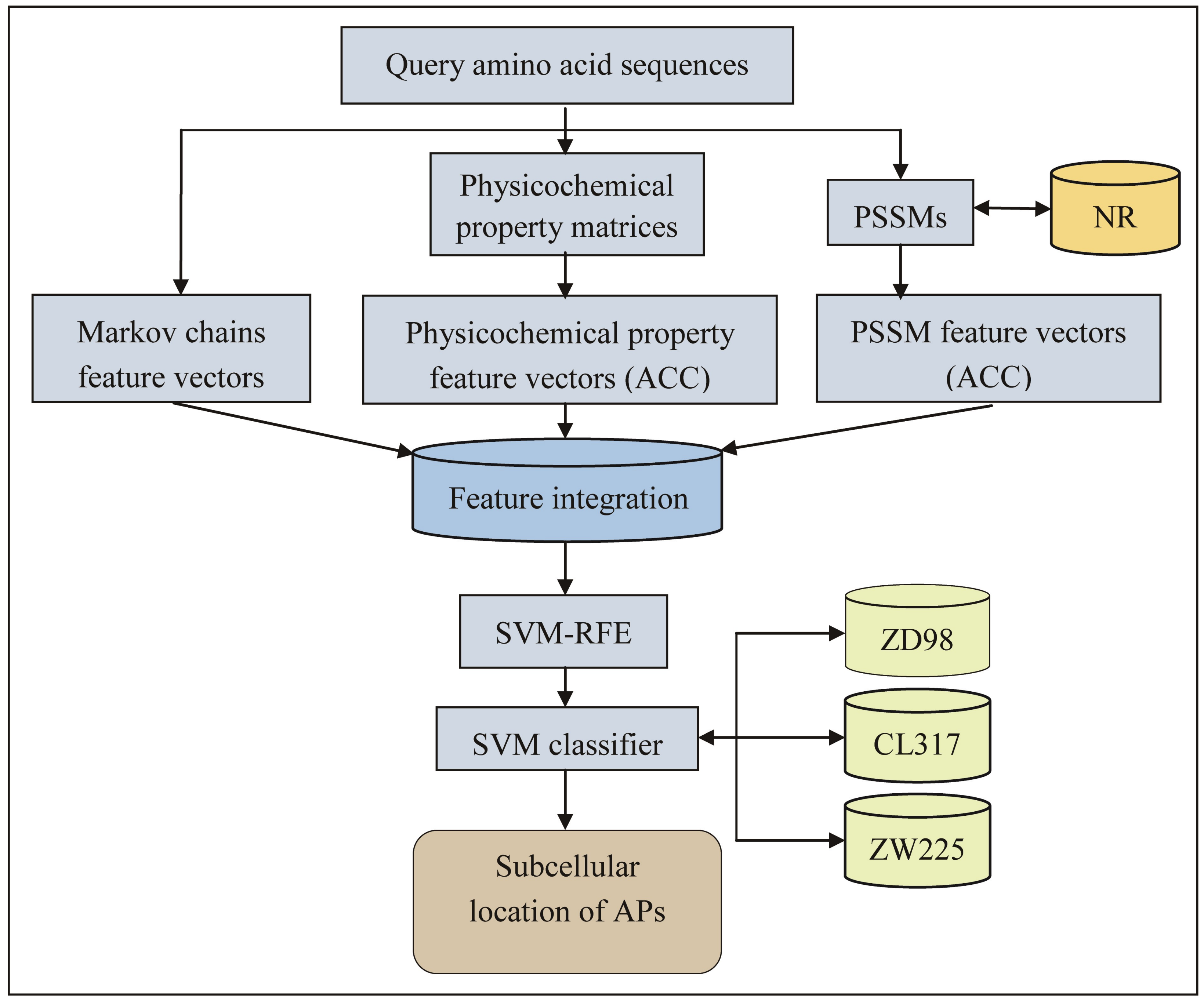
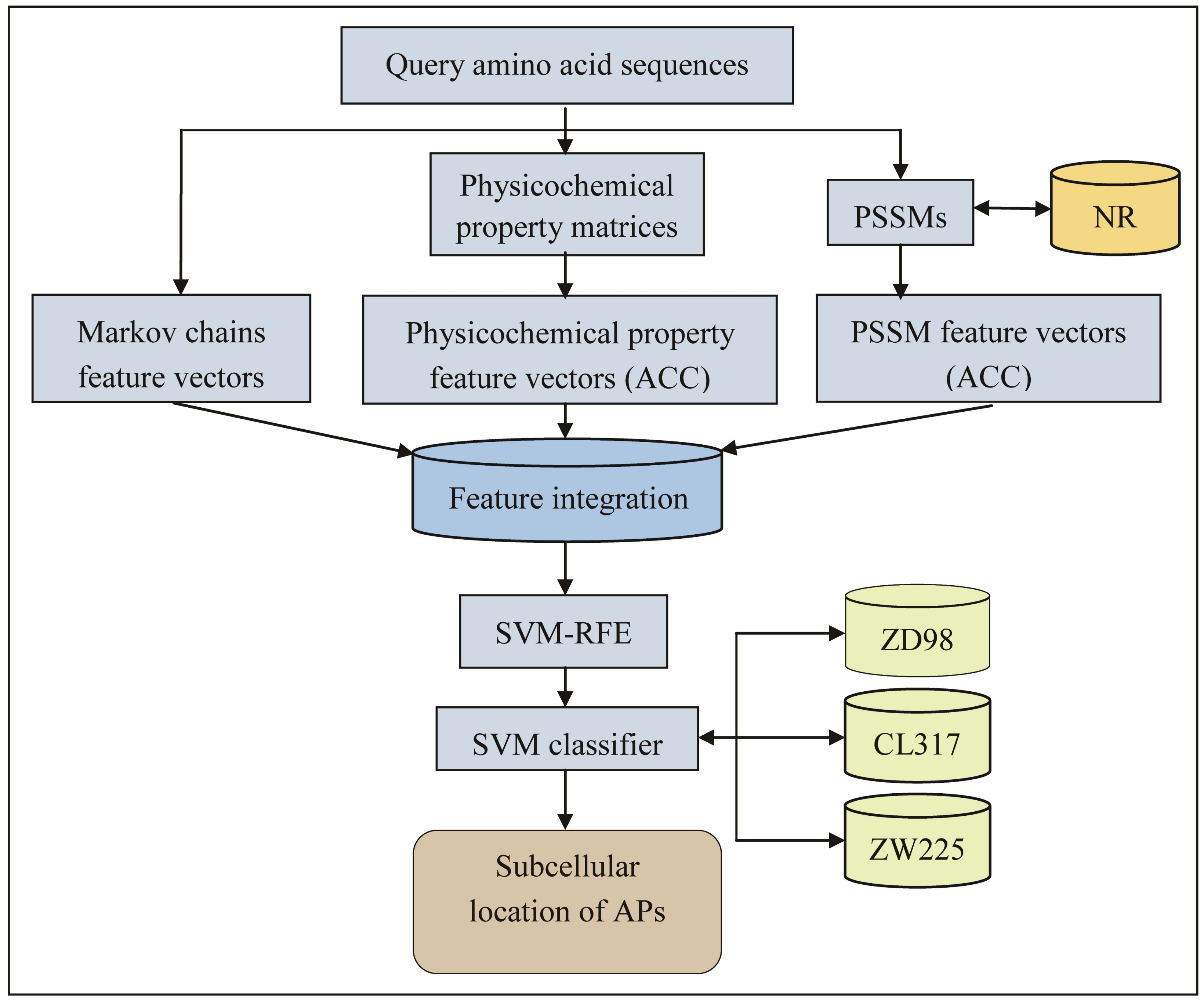
2.1. Feature Extraction
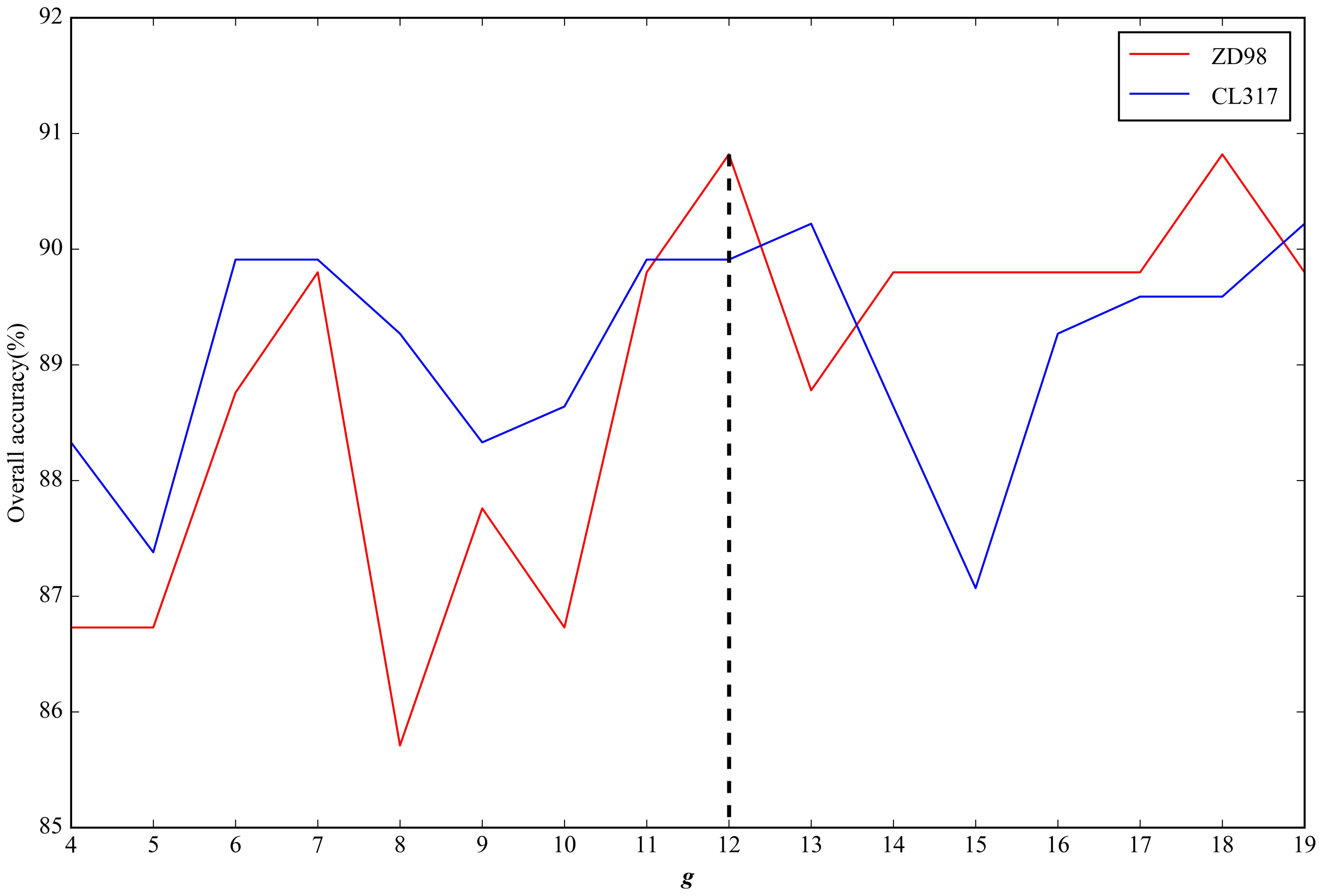
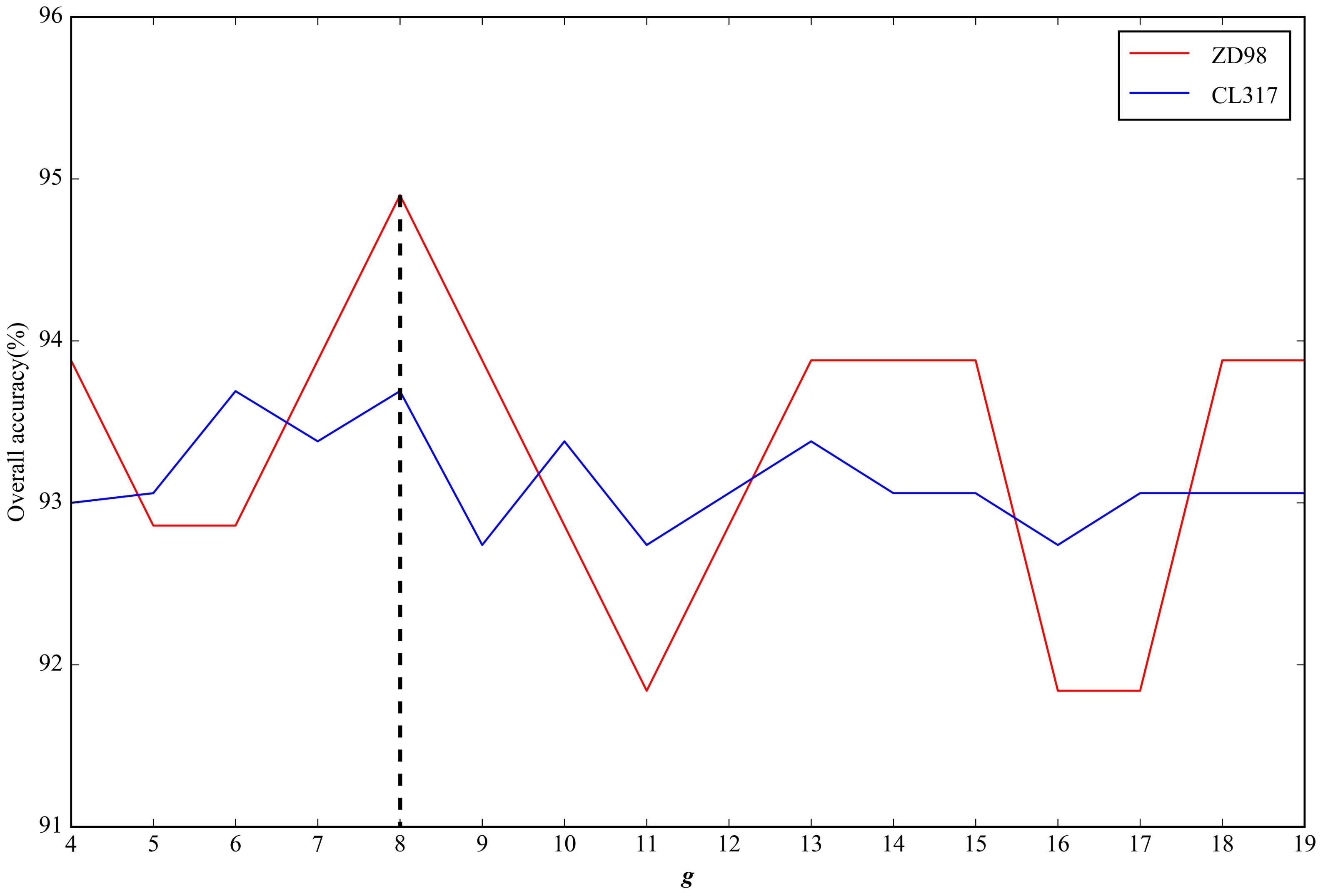
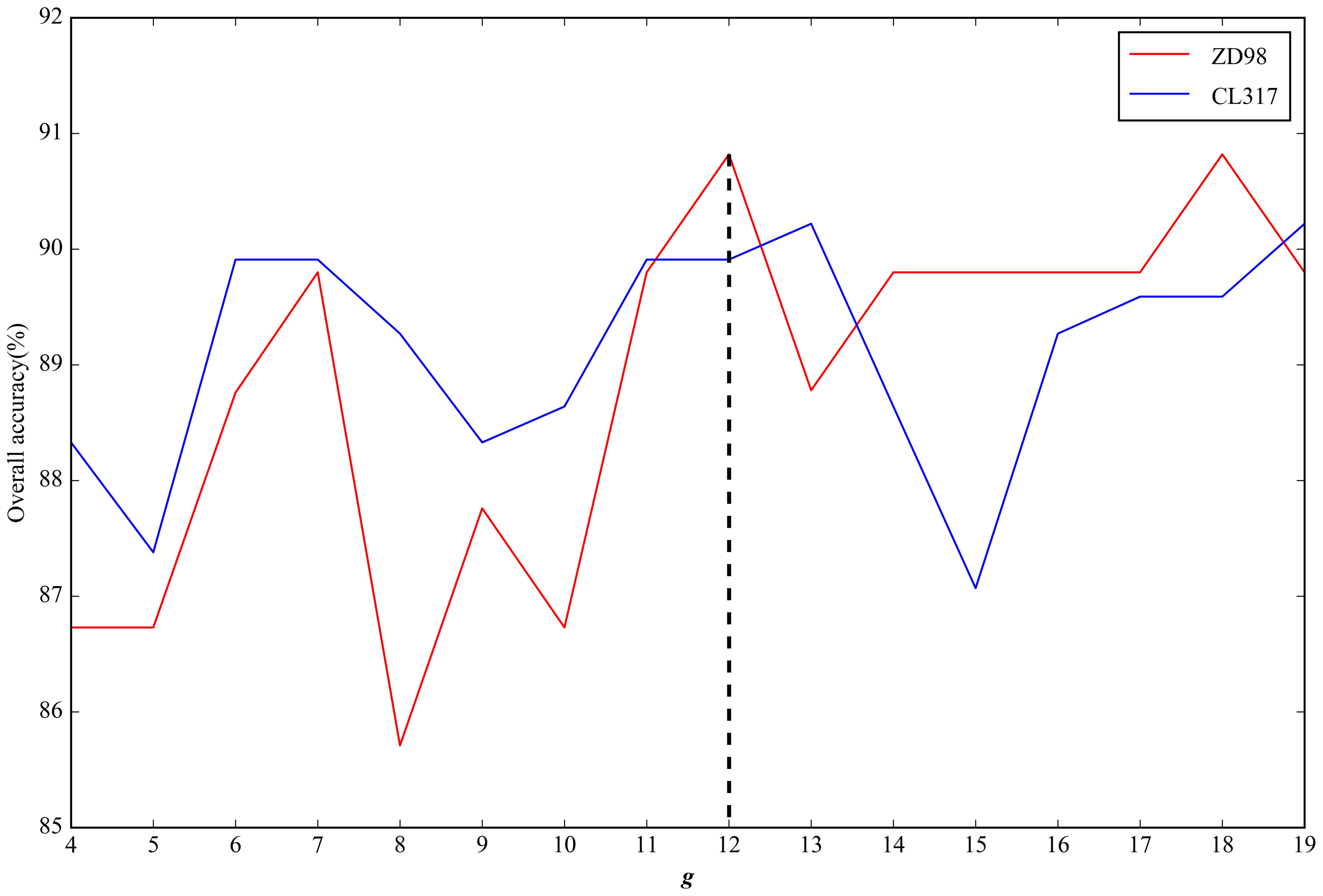
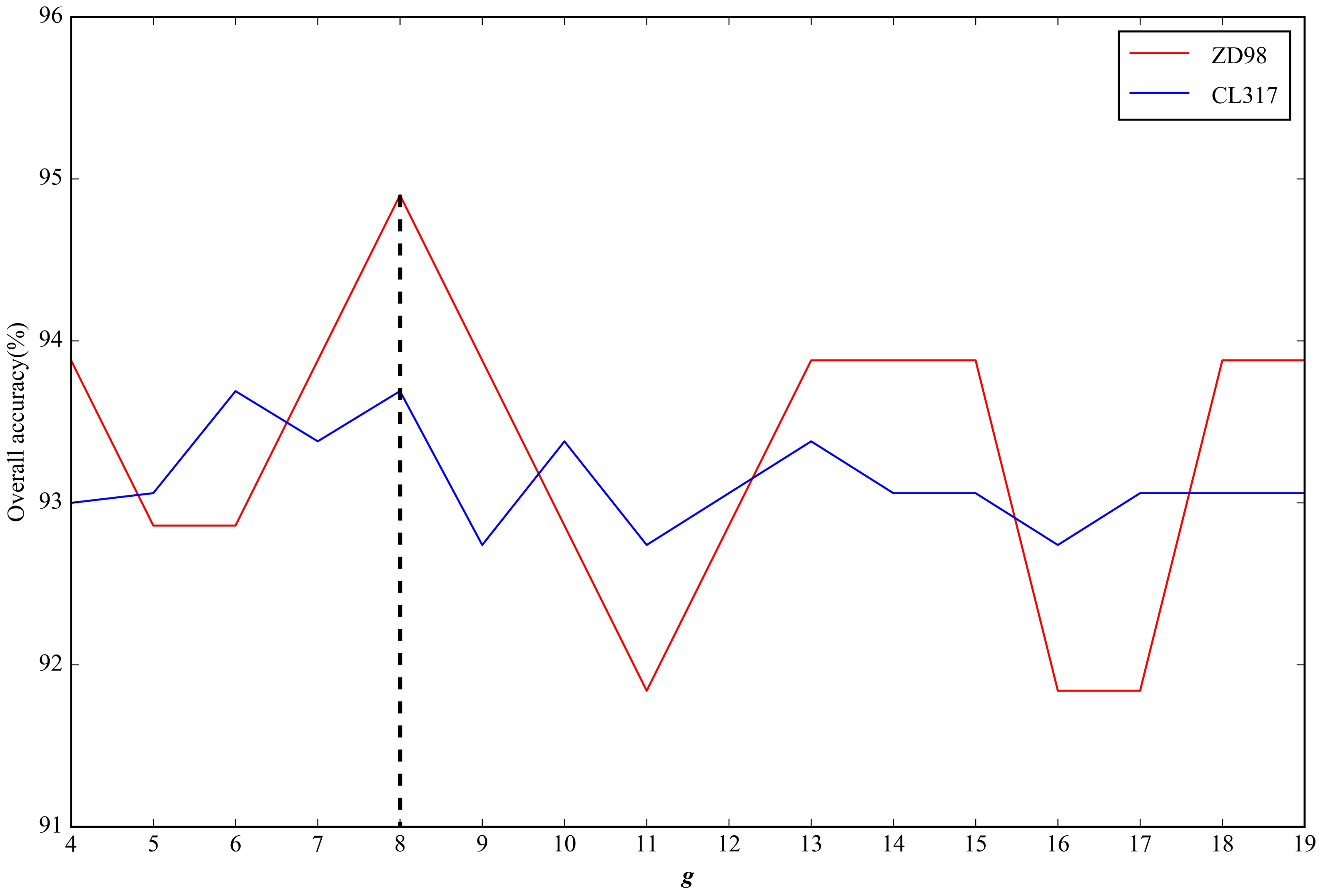
2.2. Parameter Selection
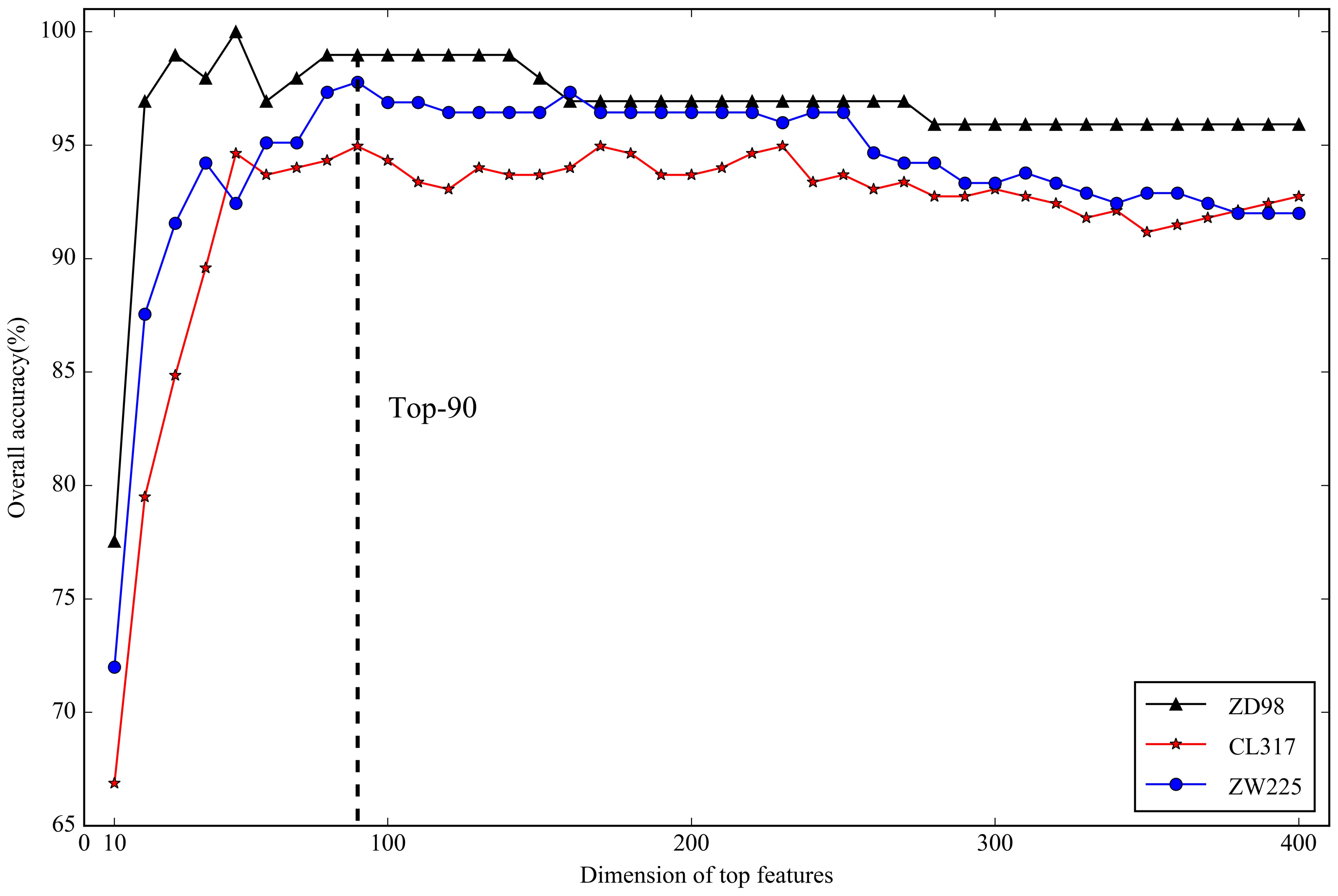
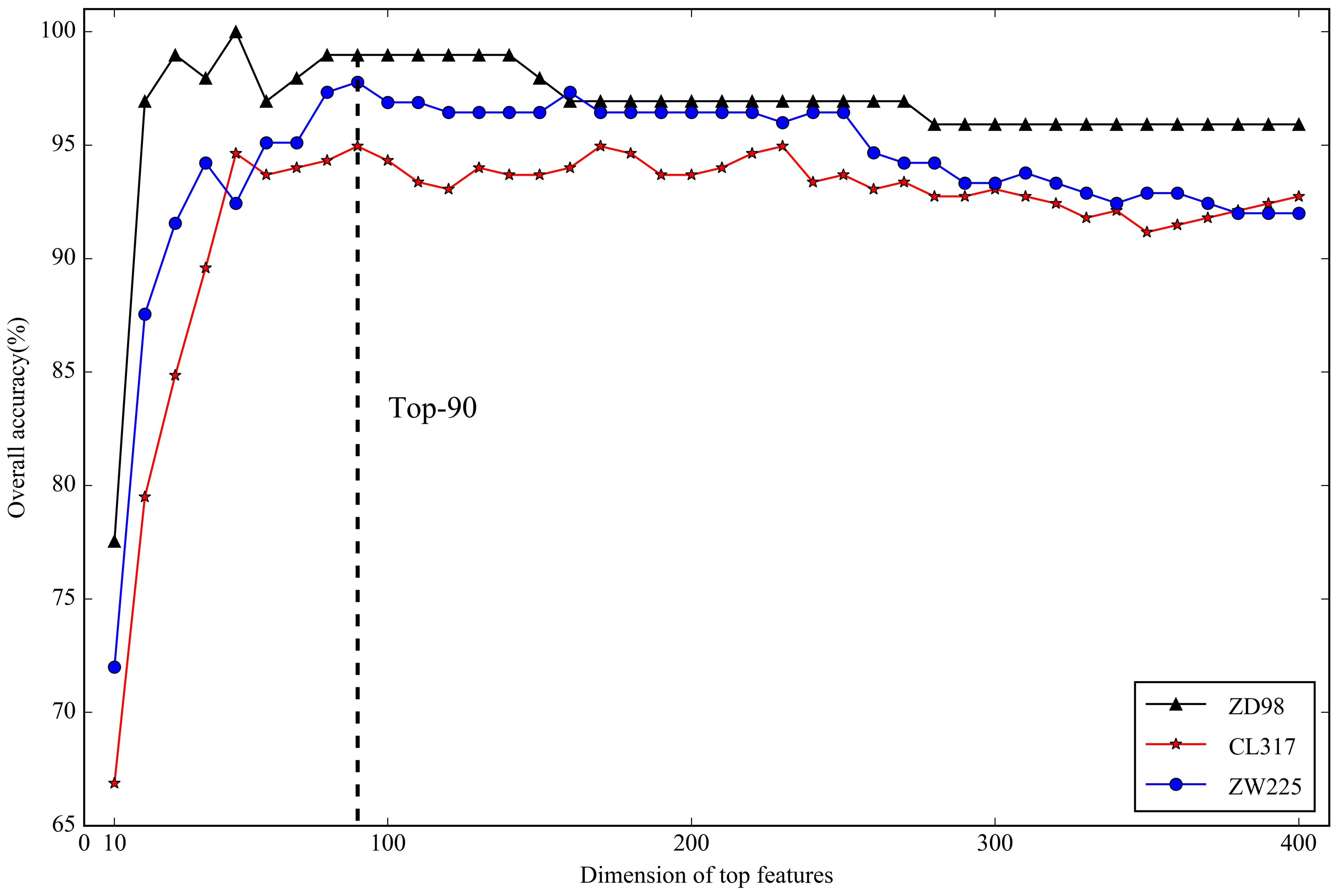
2.3. Optimal Feature Selection
2.4. Performance of the iAPSL-IF
2.5. Performance Comparison with Other Known Methods
3. Materials and Methods
3.1. Datasets
3.2. Markov Chains
3.3. Physiochemical Properties of Amino Acids
3.4. PSSM
3.5. ACC Transformation
3.6. SVM and SVM-RFE
3.7. Performance Measurement
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guo, P.Z.; Kutbuddin, D. Subcellular Location prediction of apoptosis proteins. Protein Struct. Funct. Genet. 2003, 50, 44–48. [Google Scholar]
- Zhang, Z.H.; Wang, Z.H.; Zhang, Z.R.; Wang, Y.X. A novel method for apoptosis protein subcellular localization prediction combining encoding based on grouped weight and support vector machine. FEBS Lett. 2006, 580, 6169–6174. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.D.; Luo, S.H.; Huang, J.H.; Sun, X.Y.; Liang, R.P. Predicting subcellular location of apoptosis proteins based on wavelet transform and support vector machine. Amino Acids 2010, 38, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Orrenius, S.; Zhivotovsky, B. Apoptosis in human disease: A new skin for the old ceremony? Biochem. Biophys. Res. Commun. 1999, 266, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, F.; Ju, Y.; Wang, Z.; Wang, C. Human protein subcellular localization with integrated source and multi-label ensemble classifier. Sci. Rep. 2016, 6, 28087. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Wang, X. DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation. Sci. Rep. 2015, 5, 15479. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tao, P.; Li, X.; Qin, Y.; Wang, C. Prediction of subcellular location of apoptosis proteins combining tri-gram encoding based on pssm and recursive feature elimination. J. Theor. Biol. 2015, 366, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.M.; Weng, S.L.; Lu, C.T.; Chang, T.H.; Weng, J.T.; Lee, T.Y. Sohsite: Incorporating evolutionary information and physicochemical properties to identify protein s-sulfenylation sites. BMC Genom. 2016, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Kanehisa, M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 2003, 19, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Li, Q.Z. Prediction of apoptosis protein subcellular location using improved hybrid approach and pseudo-amino acid composition. J. Theor. Biol. 2007, 248, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Li, Q.Z. Prediction of the subcellular location of apoptosis proteins. J. Theor. Biol. 2007, 245, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-S.; Zhang, T.-L. Using Chou’s pseudo amino acid composition to predict subcellular localization of apoptosis proteins: An approach with immune genetic algorithm-based ensemble classifier. Pattern Recognit. Lett. 2008, 29, 1887–1892. [Google Scholar] [CrossRef]
- Liao, B.; Jiang, J.B.; Zeng, Q.G.; Zhu, W. Predicting apoptosis protein subcellular location with PseAAC by incorporating tripeptide composition. Protein Peptide Lett. 2011, 18, 1086–1092. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, X.; Liu, T.; Dou, Y.; Wang, J. Predicting subcellular location of apoptosis proteins with pseudo amino acid composition: Approach from amino acid substitution matrix and auto covariance transformation. Amino Acids 2012, 42, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Bulashevska, A.; Eils, R. Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinform. 2006, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liao, B.; Li, D.; Zhu, W. A novel representation for apoptosis protein subcellular localization prediction using support vector machine. J. Theor. Biol. 2009, 259, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Liu, S.; Zhang, S. Detrended cross-correlation coefficient: Application to predict apoptosis protein subcellular localization. Math. Biosci. 2016, 282, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Liao, B.; Li, X.; Xu, H.; Chen, J.; Shi, Z.; Dai, Q.; Yao, Y. Subcellular localization prediction of apoptosis proteins based on evolutionary information and support vector machine. Artif. Intell. Med. 2017, 78, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Paternostro, G. Postmitochomdrial regulation of apoptosis during heart failure. Proc. Natl. Acad. Sci. USA 1999, 96, 7614–7616. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of bax: Coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef]
- Wang, T.; Yun, J.H.; Xie, Y.; Xiao, G.H. Finding RNA-protein interaction sites using HMMs. Methods Mol. Biol. 2017, 1552, 177–184. [Google Scholar] [PubMed]
- Yun, J.Y.; Wang, T.; Xiao, G.H. Bayesian Hidden Markov Models to identify RNA-protein interaction sites in PAR-CLIP. Biometrics 2014, 70, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Qin, Y.; Wang, Y.; Wang, C. Prediction of protein structural class based on gapped-dipeptides and a recursive feature selection approach. Int. J. Mol. Sci. 2016, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liao, M.; Gao, X.; Wang, J.; Lin, W. Mgof-loc: A novel ensemble learning method for human protein subcellular localization prediction. Neurocomputing 2016, 217, 73–82. [Google Scholar] [CrossRef]
- Tao, P.; Liu, T.; Li, X.; Chen, L. Prediction of protein structural class using tri-gram probabilities of position-specific scoring matrix and recursive feature elimination. Amino Acids 2015, 47, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Ding, Y.S.; Jiang, X.Y.; Zhang, T.L. Prediction of subcellular location apoptosis proteins with ensemble classifier and feature selection. Amino Acids 2010, 38, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, V.; Lakshmi, P.T. Apslap: An adaptive boosting technique for predicting subcellular localization of apoptosis protein. Acta Biotheor. 2013, 61, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Vapnik, V. Statistical Learning Theory; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Wang, Y.; Ding, Y.; Guo, F.; Wei, L.; Tang, J. Improved detection of DNA-binding proteins via compression technology on PSSM information. PLoS ONE 2017, 12, e0185587. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Jonsson, J.; Sjostrom, M.; Sandberg, M.; Rannar, S. DNA and peptide sequences and chemical processes multivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal. Chim. Acta 1993, 277, 239–253. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, L.; Dong, Q. Protein remote homology detection based on auto-cross covariance transformation. Comput. Biol. Med. 2011, 41, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, T.; Tao, P.; Wang, C.; Chen, L. A highly accurate protein structural class prediction approach using auto cross covariance transformation and recursive feature elimination. Comput. Biol. Chem. 2015, 59 Pt A, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Papini, A.; Kim Rossmo, D.; Le Comber, S.C.; Verity, R.; Stevenson, M.D.; Santosuosso, U. The use of jackknifing for the evaluation of geographic profiling reliability. Ecol. Inform. 2017, 38, 76–81. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Wang, R.; Zhang, Q.; Zhang, W.; Gan, Y. Multip-apo: A multilabel predictor for identifying subcellular locations of apoptosis proteins. Comput. Intell. Neurosci. 2017, 2017, 9183796. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Weston, J.; Barnhill, S. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Lin, H.; Wang, H.; Ding, H.; Chen, Y.L.; Li, Q.Z. Prediction of subcellular localization of apoptosis protein using chou’s pseudo amino acid composition. Acta Biotheor. 2009, 57, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.L.; Zou, Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Improved prediction of subcellular location for apoptosis proteins by the dual-layer support vector machine. Amino Acids 2008, 35, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Banchariya, A.; Rao, A.R. Dirprot: A computational approach for discriminating insecticide resistant proteins from non-resistant proteins. BMC Bioinform. 2017, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Hayat, M. Machine learning based identification of protein-protein interactions using derived features of physiochemical properties and evolutionary profiles. Artif. Intell. Med. 2017, 78, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iPPI-Esml: An ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J. Theor. Biol. 2015, 377, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, B. Psfm-dbt: Identifying DNA-binding proteins by combing position specific frequency matrix and distance-bigram transformation. Int. J. Mol. Sci. 2017, 18, 1856. [Google Scholar]
- Liu, B.; Wang, S.; Dong, Q.; Li, S.; Liu, X. Identification of DNA-binding proteins by combining auto-cross covariance transformation and ensemble learning. IEEE Trans. Nanobiosci. 2016, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Liu, T.; Geng, X.; Zheng, X.; Li, R.; Wang, J. Accurate prediction of protein structural class using auto covariance transformation of psi-blast profiles. Amino Acids 2012, 42, 2243–2249. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Location | Sens (%) | Spec (%) | MCC | OA (%) |
|---|---|---|---|---|---|
| ZD98 | Cyto | 97.67 | 100 | 0.98 | 98.98 |
| Memb | 100 | 98.53 | 0.98 | ||
| Mito | 100 | 100 | 1.0 | ||
| other | 100 | 100 | 1.0 | ||
| CL317 | Cyto | 95.54 | 97.07 | 0.92 | 94.95 |
| Memb | 94.55 | 98.85 | 0.93 | ||
| Mito | 88.24 | 98.94 | 0.88 | ||
| Secr | 100 | 99.67 | 0.97 | ||
| Nucl | 94.23 | 98.87 | 0.93 | ||
| Endo | 97.87 | 100 | 0.99 | ||
| ZW225 | Cyto | 100 | 98.71 | 0.98 | 97.33 |
| Memb | 98.88 | 99.26 | 0.98 | ||
| Mito | 88.00 | 99.50 | 0.90 | ||
| Nucl | 95.12 | 98.98 | 0.94 |
| Method | Sens for Each Class (%) | OA (%) | Reference | |||
|---|---|---|---|---|---|---|
| Cyto | Memb | Mito | Other | |||
| Covariant | 97.7 | 73.3 | 30.8 | 25.0 | 72.5 | [1] |
| ID_SVM | 95.3 | 93.3 | 84.6 | 58.3 | 88.8 | [10] |
| DWT_SVM | 95.4 | 93.3 | 53.9 | 91.7 | 88.8 | [26] |
| ID | 90.7 | 90.0 | 92.3 | 91.7 | 90.8 | [11] |
| EBGW_SVM | 97.7 | 90.0 | 92.3 | 83.3 | 92.9 | [2] |
| PseAAC_SVM | 95.3 | 93.3 | 92.3 | 83.3 | 92.9 | [36] |
| DF_SVM | 97.7 | 96.7 | 92.3 | 75.0 | 93.9 | [16] |
| Dual_layer SVM | 95.4 | 96.7 | 92.3 | 91.7 | 94.9 | [37] |
| APSLAP | 95.3 | 90.0 | 100 | 91.7 | 94.9 | [27] |
| FKNN | 95.3 | 96.7 | 100 | 91.7 | 95.9 | [12] |
| PSSM-AC | 97.7 | 96.7 | 100 | 83.3 | 95.9 | [38] |
| PSSM-trigram | 95.3 | 100 | 100 | 91.7 | 96.9 | [7] |
| iAPSL-IF | 97.7 | 100 | 100 | 100 | 99.0 | This study |
| Method | Sens for Each Class (%) | OA (%) | Reference | |||||
|---|---|---|---|---|---|---|---|---|
| Cyto | Memb | Mito | Secr | Nucl | Endo | |||
| ID | 81.3 | 81.8 | 85.3 | 88.2 | 82.7 | 83.0 | 82.7 | [10] |
| ID_SVM | 91.1 | 89.1 | 79.4 | 58.8 | 73.1 | 87.2 | 84.2 | [11] |
| DF_SVM | 92.9 | 85.5 | 76.5 | 76.5 | 93.6 | 86.5 | 88.0 | [16] |
| Auto_Cova | 86.4 | 90.7 | 93.8 | 85.7 | 92.1 | 93.8 | 90.0 | [14] |
| FKNN | 93.8 | 92.7 | 82.4 | 76.5 | 90.4 | 93.6 | 90.9 | [12] |
| PseAAC_SVM | 93.8 | 90.9 | 85.3 | 76.5 | 90.4 | 95.7 | 91.1 | [36] |
| EN_FKNN | 98.2 | 83.6 | 79.4 | 82.4 | 90.4 | 97.9 | 91.5 | [26] |
| PSSM-AC | 93.8 | 90.9 | 91.2 | 82.4 | 86.5 | 95.7 | 91.5 | [38] |
| APSLAP | 99.1 | 89.1 | 85.3 | 88.2 | 84.3 | 95.8 | 92.4 | [27] |
| EI_SVM | 94.6 | 95.7 | 92.7 | 82.4 | 90.4 | 70.6 | 91.1 | [18] |
| iAPSL-IF | 95.5 | 94.5 | 88.2 | 100 | 94.2 | 97.9 | 95.0 | This study |
| Method | Sens for Each Class (%) | OA (%) | Reference | |||
|---|---|---|---|---|---|---|
| Cyto | Memb | Mito | Nucl | |||
| EBGW_SVM | 90.0 | 93.3 | 60.0 | 63.4 | 83.1 | [2] |
| DF_SVM | 87.1 | 92.1 | 64.0 | 73.2 | 84.0 | [16] |
| PSSM-AC | 82.9 | 92.1 | 68.0 | 78.0 | 84.0 | [38] |
| ID_SVM | 92.9 | 91.0 | 68.0 | 73.2 | 85.8 | [11] |
| Auto_Cova | 81.3 | 93.3 | 85.7 | 84.6 | 87.1 | [14] |
| EN_FKNN | 94.3 | 94.4 | 60.0 | 80.5 | 88.0 | [26] |
| PSSM-trigram | 97.1 | 98.9 | 96.0 | 97.6 | 97.8 | [7] |
| iAPSL-IF | 100 | 98.9 | 88.0 | 95.1 | 97.3 | This study |
| AA | P(1) | P(2) | P(3) | P(4) | P(5) | P(6) | P(7) | P(8) | P(9) | P(10) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 8.100 | −1.302 | −0.733 | 1.57 | −0.146 | 0.620 | −0.500 | 27.500 | 0.046 | 1.181 |
| C | 5.500 | 0.465 | −0.862 | −1.02 | −0.255 | 0.290 | −1.000 | 44.600 | 0.128 | 1.461 |
| D | 13.000 | 0.302 | −3.656 | −0.259 | −3.242 | −0.900 | 3.000 | 40.000 | 0.105 | 1.587 |
| E | 12.300 | −1.453 | 1.477 | 0.113 | −0.837 | −0.740 | 3.000 | 62.000 | 0.151 | 1.862 |
| F | 5.200 | −0.59 | 1.891 | −0.397 | 0.412 | 1.190 | −2.500 | 115.500 | 0.290 | 2.228 |
| G | 9.000 | 1.652 | 1.33 | 1.045 | 2.064 | 0.480 | 0.000 | 0.000 | 0.000 | 0.881 |
| H | 10.400 | −0.417 | −1.673 | −1.474 | −0.078 | −0.400 | −0.500 | 79.000 | 0.230 | 2.025 |
| I | 5.200 | −0.547 | 2.131 | 0.393 | 0.816 | 1.380 | −1.800 | 93.500 | 0.186 | 1.810 |
| K | 11.300 | −0.561 | 0.533 | −0.277 | 1.648 | −1.500 | 3.000 | 100.000 | 0.219 | 2.258 |
| L | 4.900 | −0.987 | −1.505 | 1.266 | −0.912 | 1.060 | −1.800 | 93.500 | 0.186 | 1.931 |
| M | 5.700 | −1.524 | 2.219 | −1.005 | 1.212 | 0.640 | −1.300 | 94.100 | 0.221 | 2.034 |
| N | 11.600 | 0.828 | 1.299 | −0.169 | 0.933 | −0.780 | 2.000 | 58.700 | 0.134 | 1.655 |
| P | 8.000 | 2.081 | −1.628 | 0.421 | −1.392 | 0.120 | 0.000 | 41.900 | 0.131 | 1.468 |
| Q | 10.500 | −0.179 | −3.005 | −0.503 | −1.853 | −0.850 | 0.200 | 80.700 | 0.180 | 1.932 |
| R | 10.500 | −0.055 | 1.502 | 0.44 | 2.897 | −2.530 | 3.000 | 105.000 | 0.291 | 2.560 |
| S | 9.200 | 1.399 | −4.76 | 0.67 | −2.647 | −0.180 | 0.300 | 29.300 | 0.062 | 1.298 |
| T | 8.000 | 0.326 | 2.213 | 0.908 | 1.313 | −0.050 | −0.400 | 51.300 | 0.108 | 1.525 |
| V | 5.900 | −0.279 | −0.544 | 1.242 | −1.262 | 1.080 | −1.500 | 71.500 | 0.140 | 1.645 |
| W | 5.400 | 0.009 | 0.672 | −2.128 | −0.184 | 0.810 | −3.400 | 145.500 | 0.409 | 2.663 |
| Y | 6.200 | 0.83 | 3.097 | −0.838 | 1.512 | 0.260 | −2.300 | 117.300 | 0.298 | 2.368 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Xie, L.; Chen, L. iAPSL-IF: Identification of Apoptosis Protein Subcellular Location Using Integrative Features Captured from Amino Acid Sequences. Int. J. Mol. Sci. 2018, 19, 1190. https://doi.org/10.3390/ijms19041190
Tang Y, Xie L, Chen L. iAPSL-IF: Identification of Apoptosis Protein Subcellular Location Using Integrative Features Captured from Amino Acid Sequences. International Journal of Molecular Sciences. 2018; 19(4):1190. https://doi.org/10.3390/ijms19041190
Chicago/Turabian StyleTang, Yadong, Lu Xie, and Lanming Chen. 2018. "iAPSL-IF: Identification of Apoptosis Protein Subcellular Location Using Integrative Features Captured from Amino Acid Sequences" International Journal of Molecular Sciences 19, no. 4: 1190. https://doi.org/10.3390/ijms19041190
APA StyleTang, Y., Xie, L., & Chen, L. (2018). iAPSL-IF: Identification of Apoptosis Protein Subcellular Location Using Integrative Features Captured from Amino Acid Sequences. International Journal of Molecular Sciences, 19(4), 1190. https://doi.org/10.3390/ijms19041190