Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
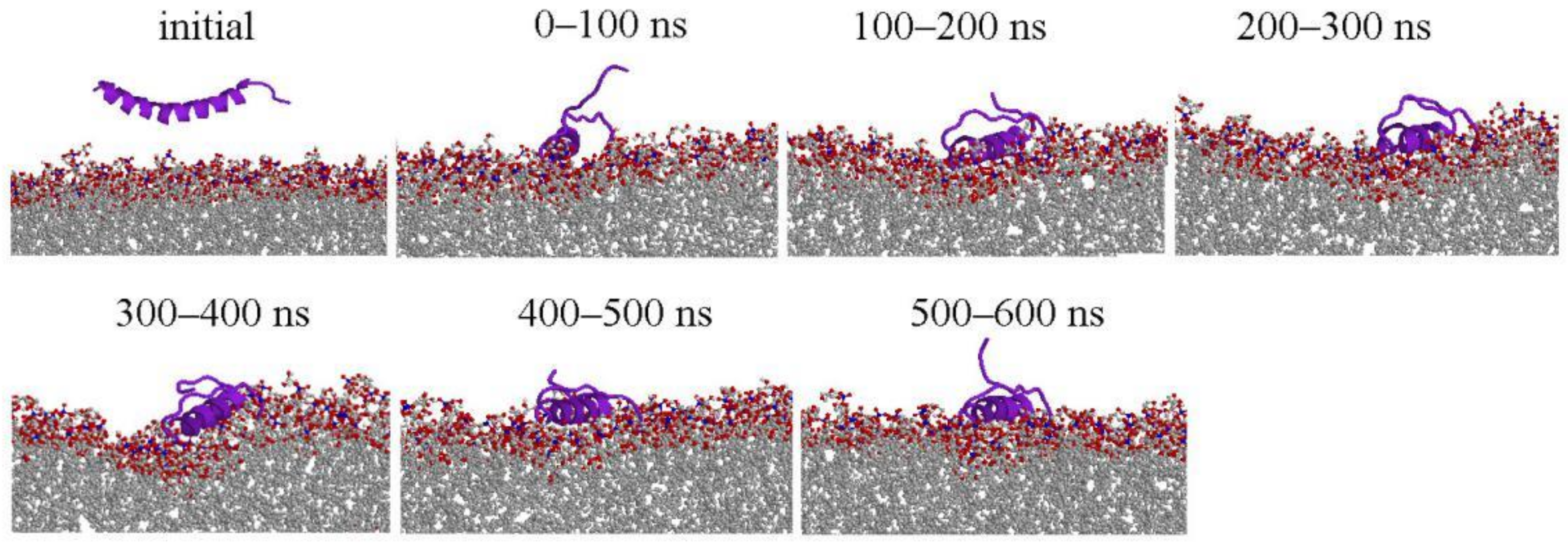
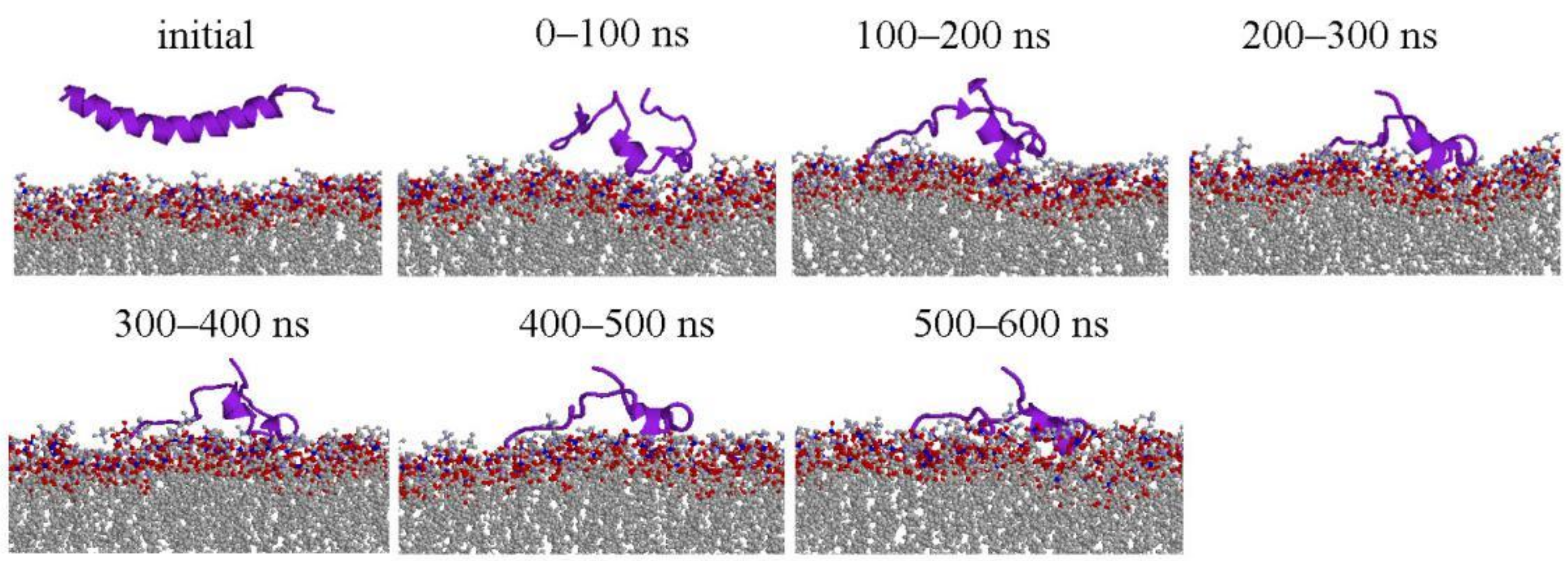
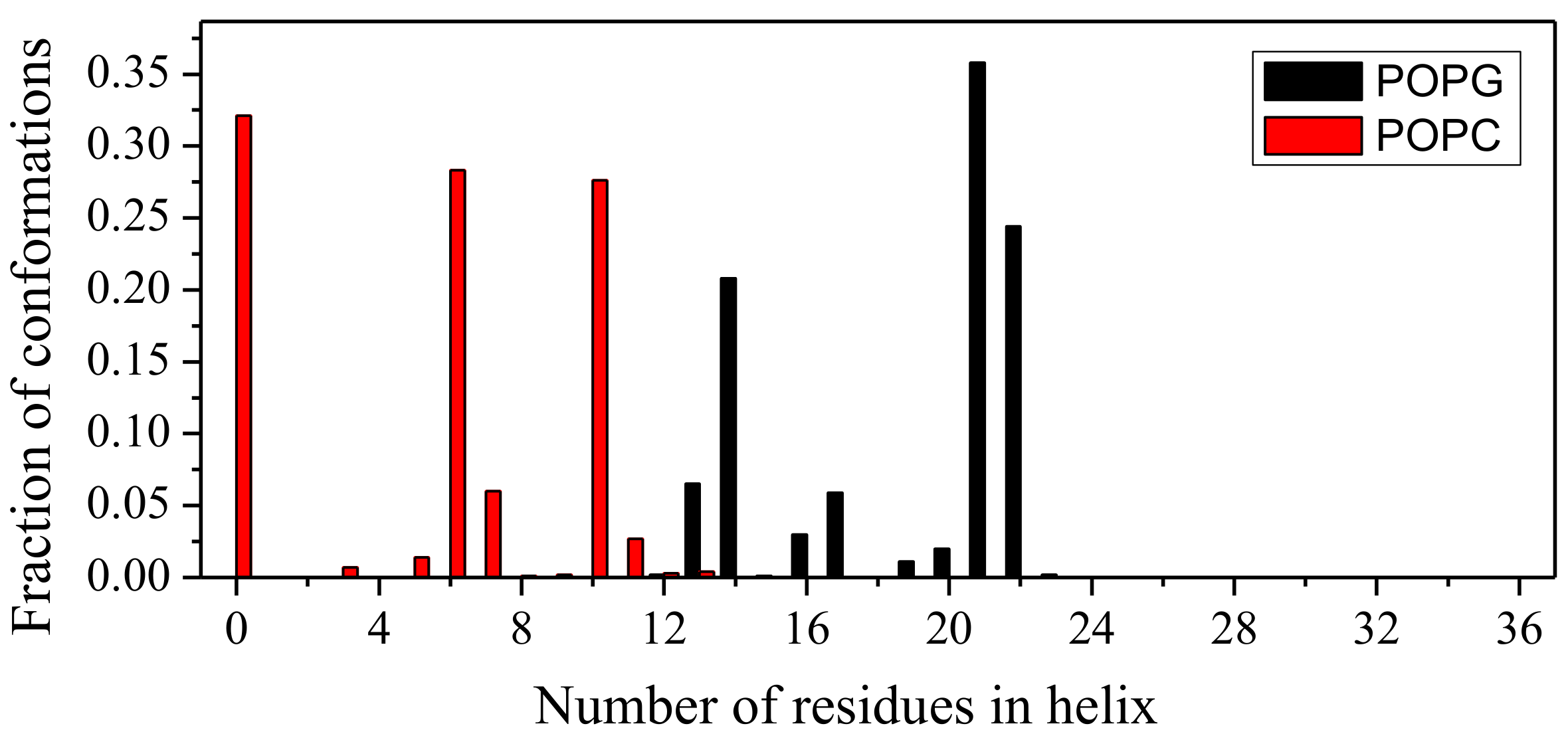
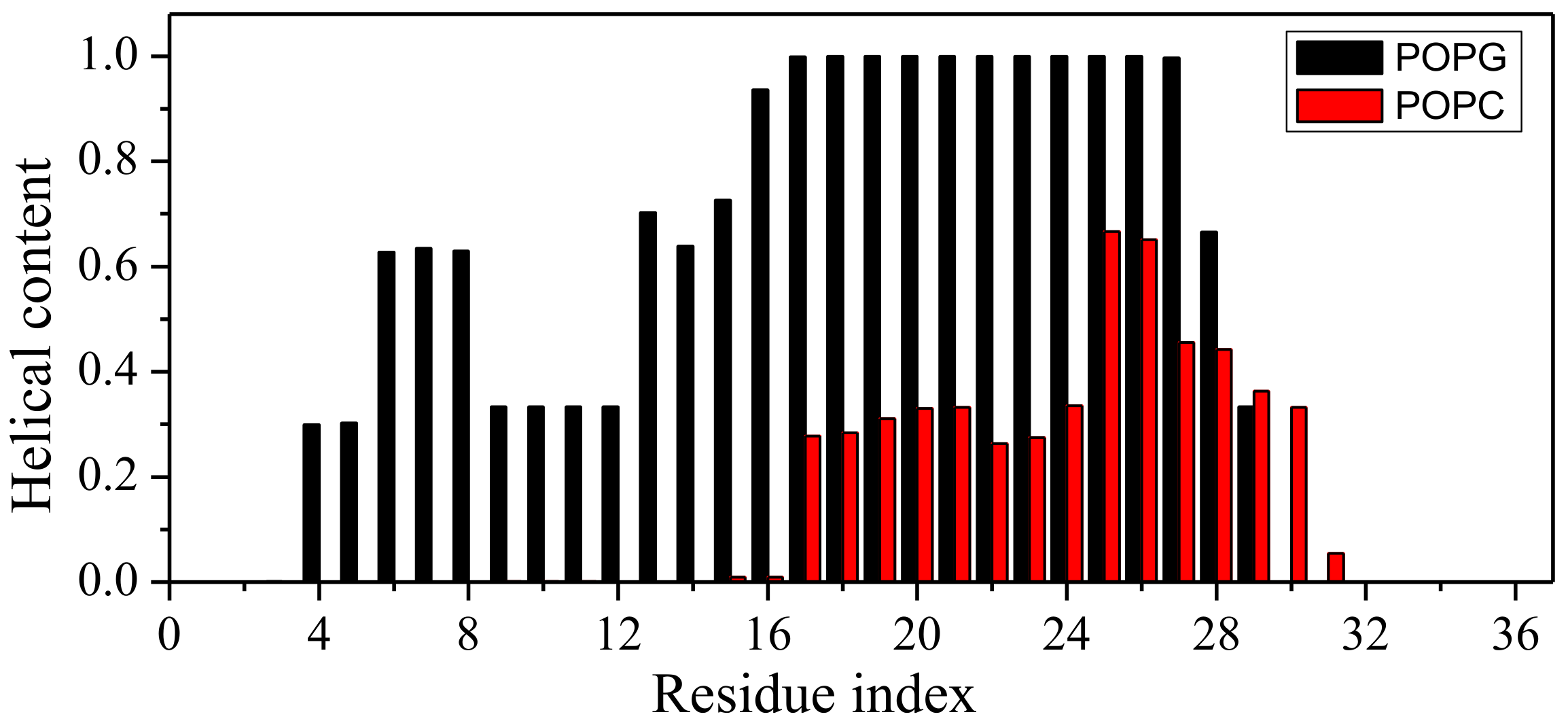
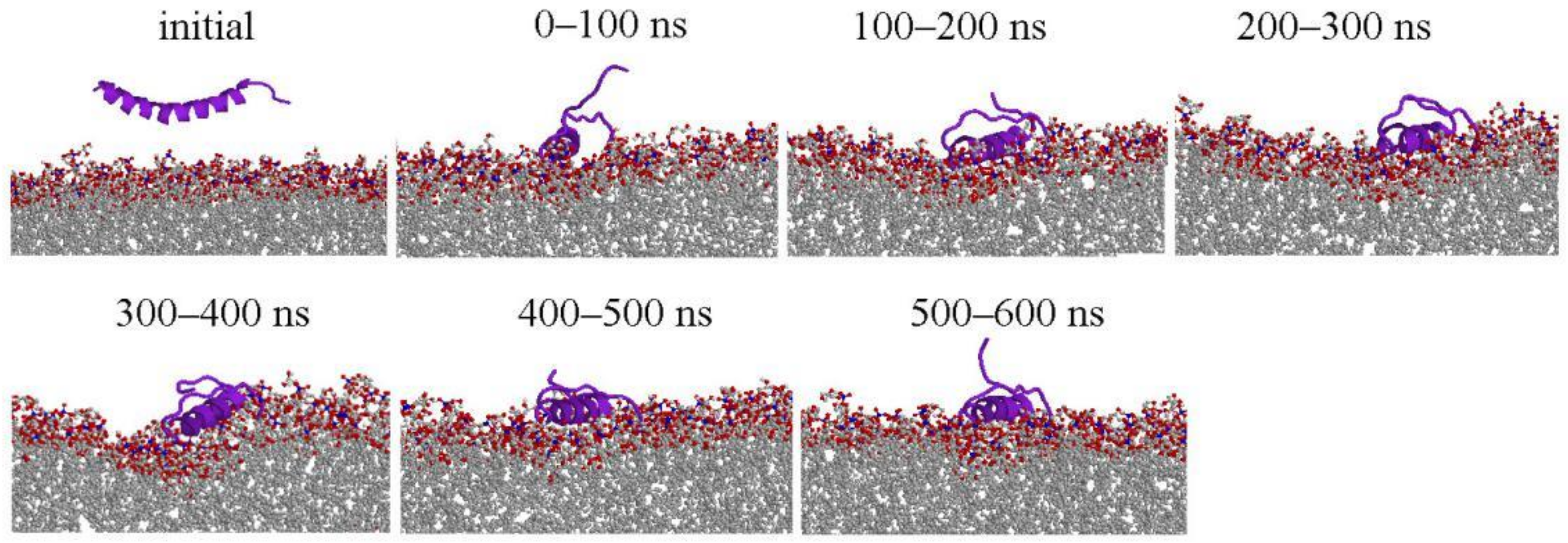
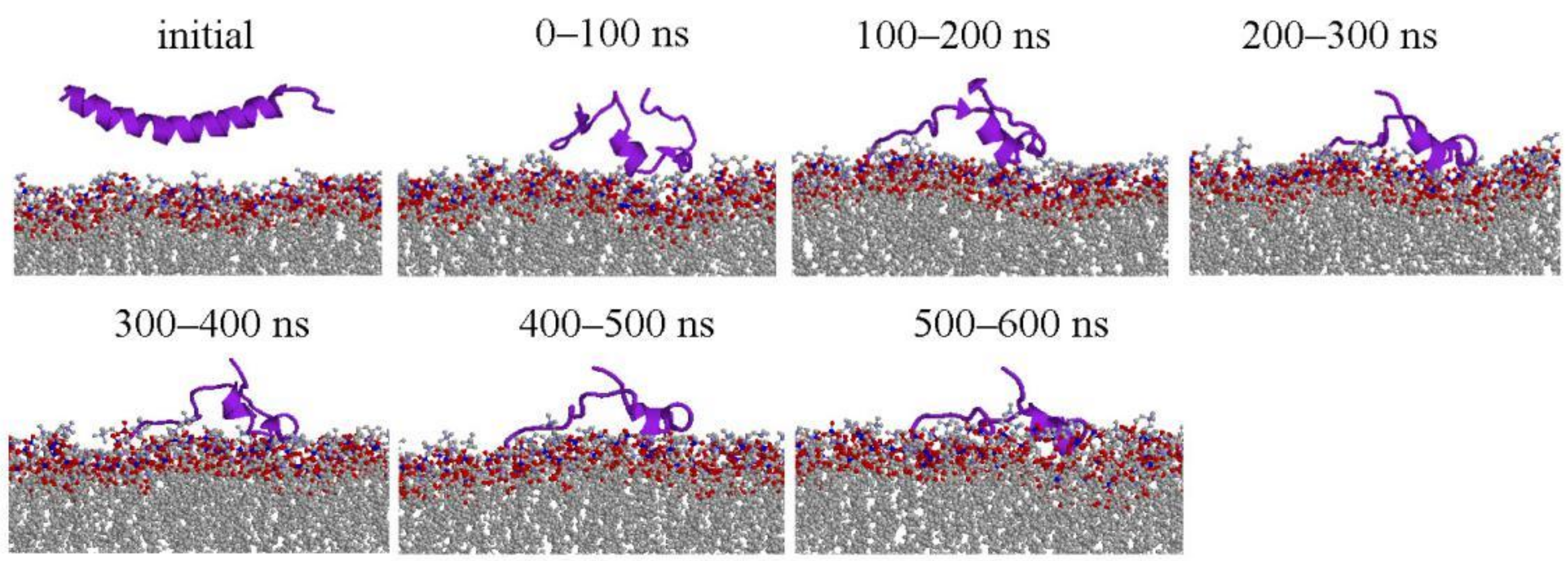
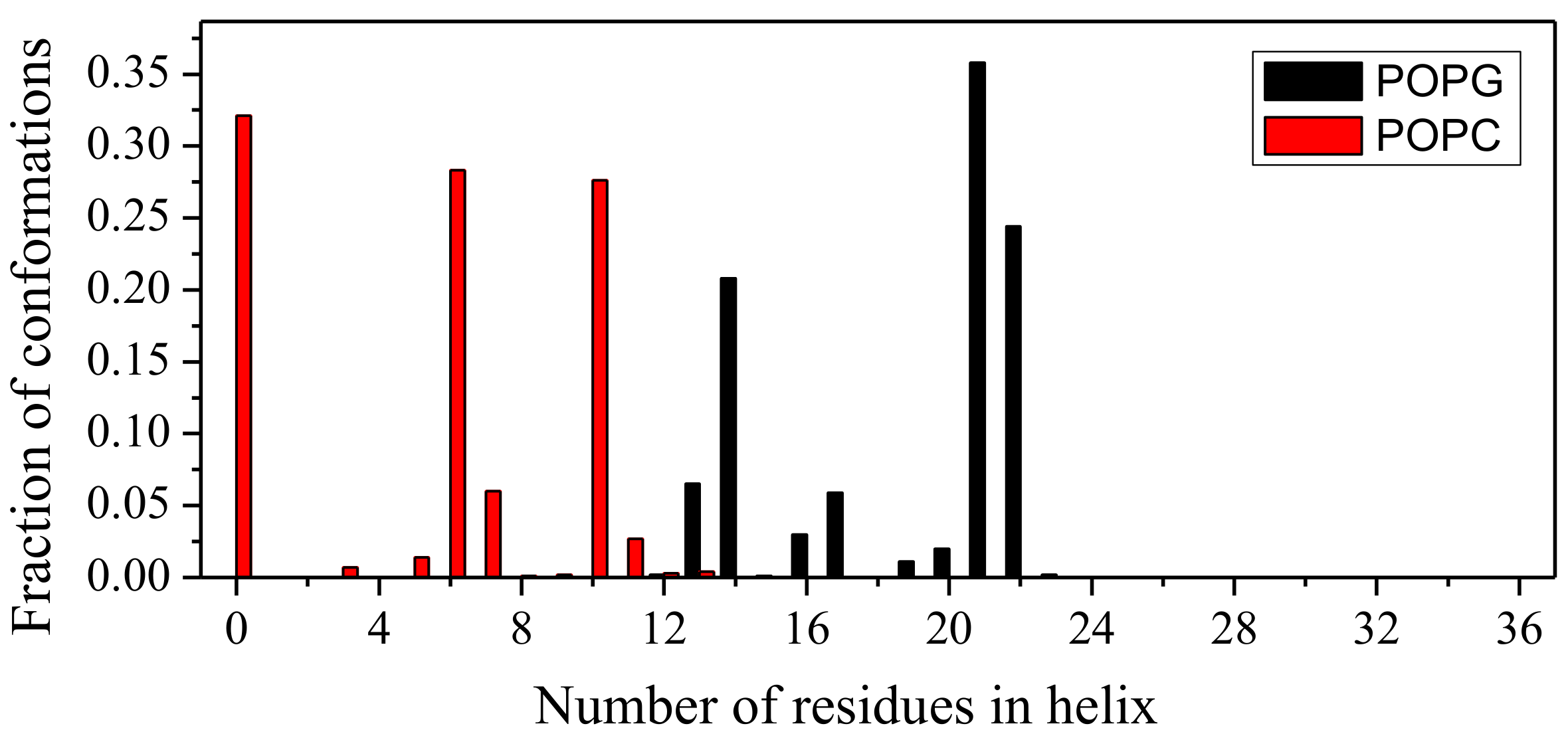
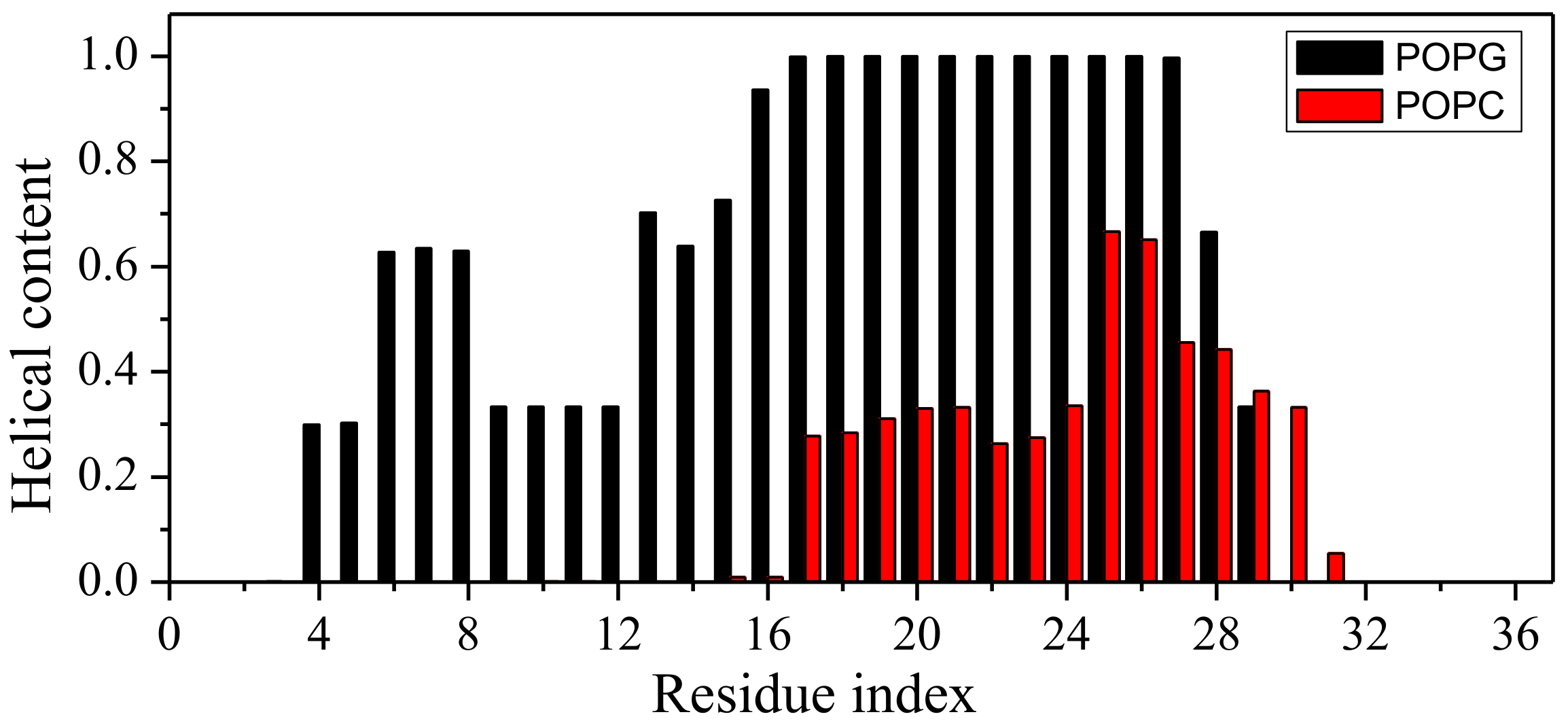
2.1. Peptide Conformations
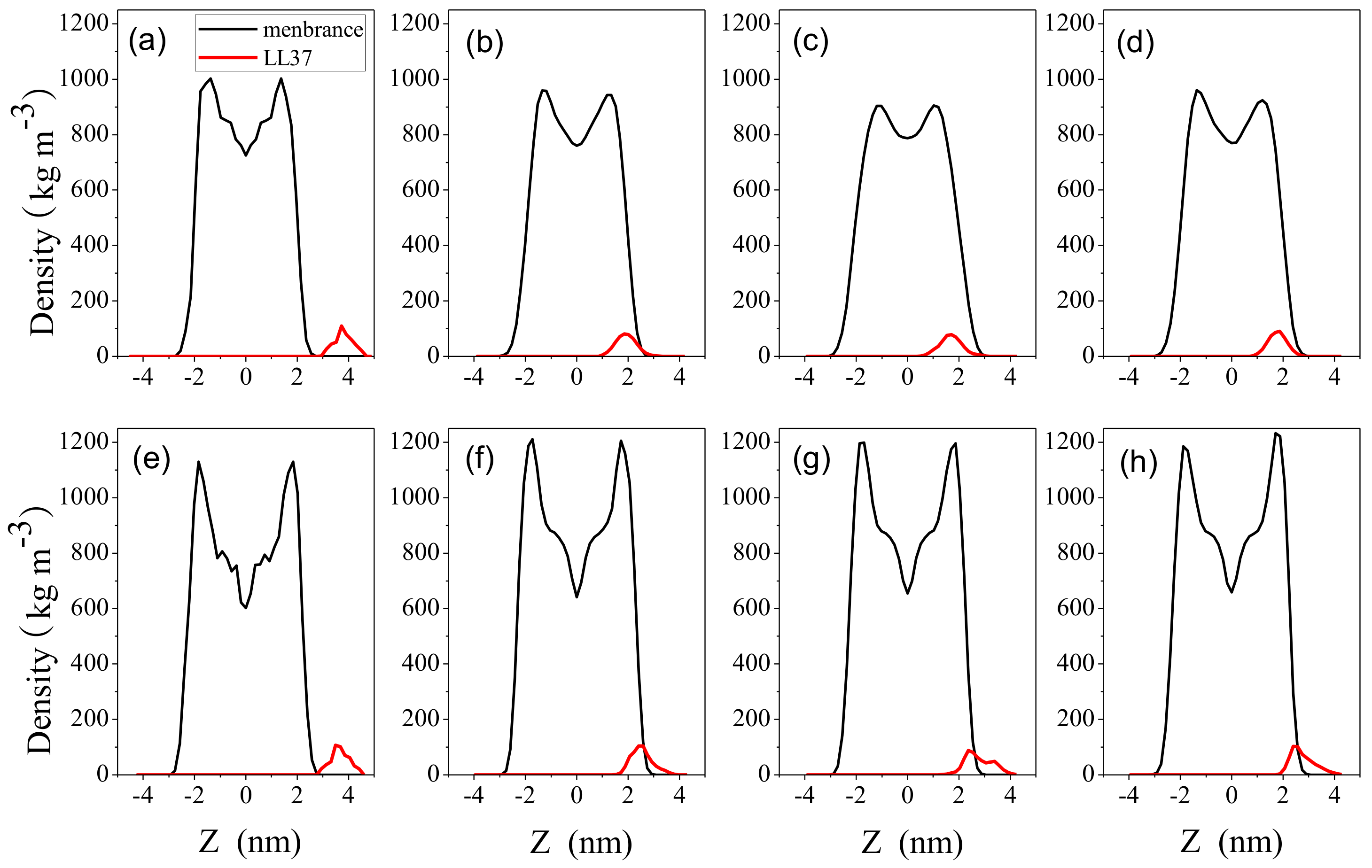
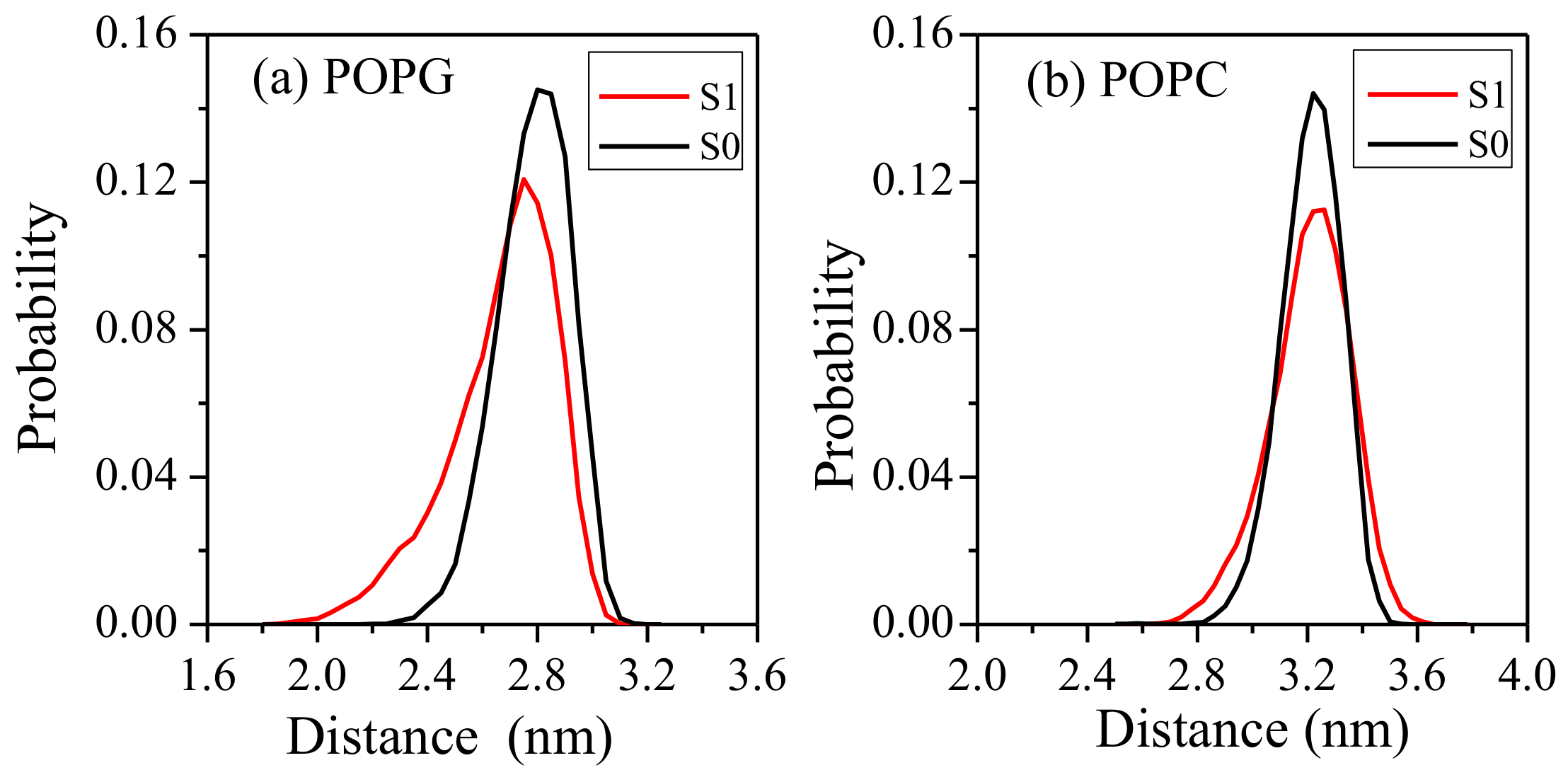
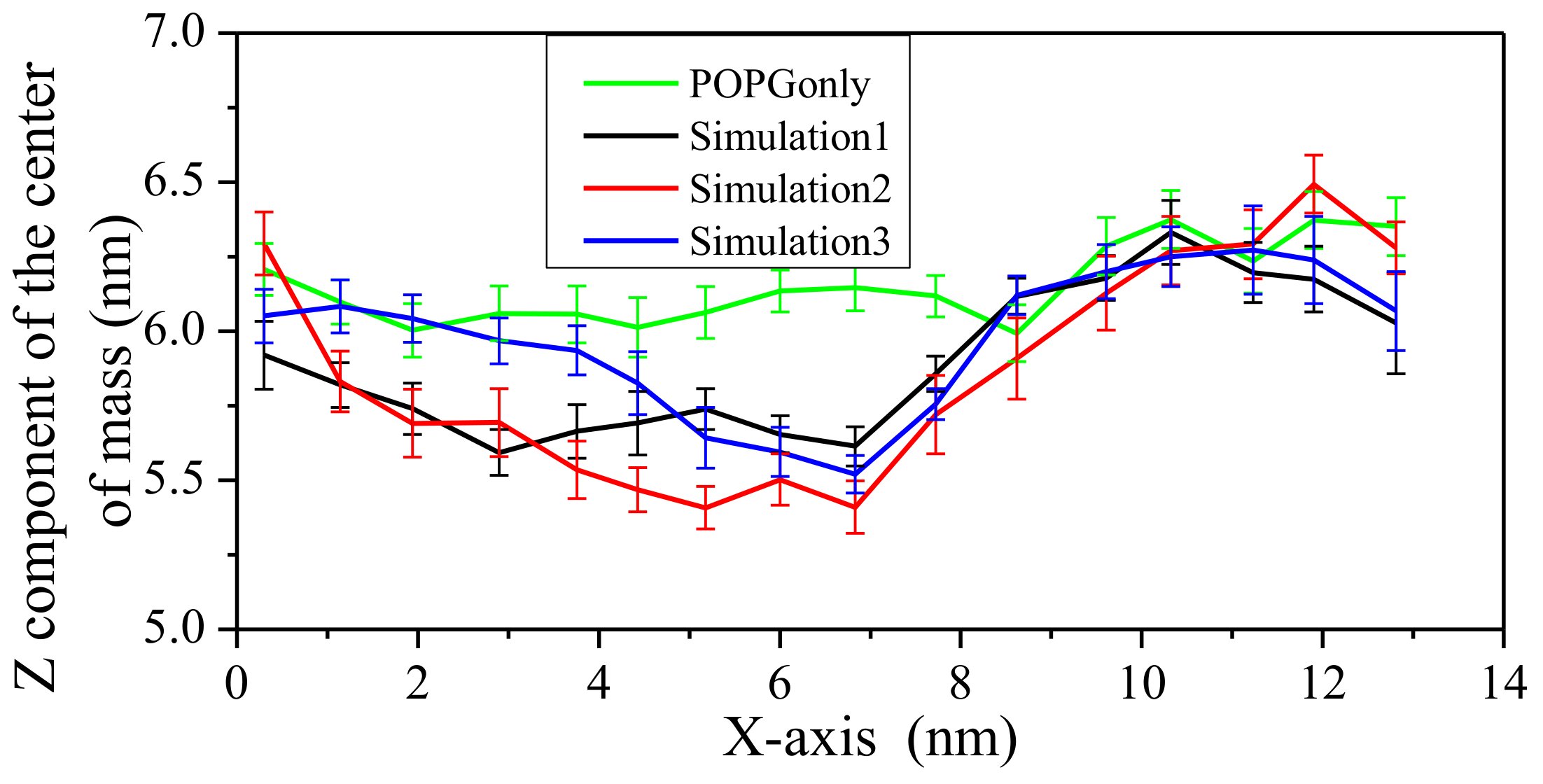
2.2. Interaction between Peptide and Membrane
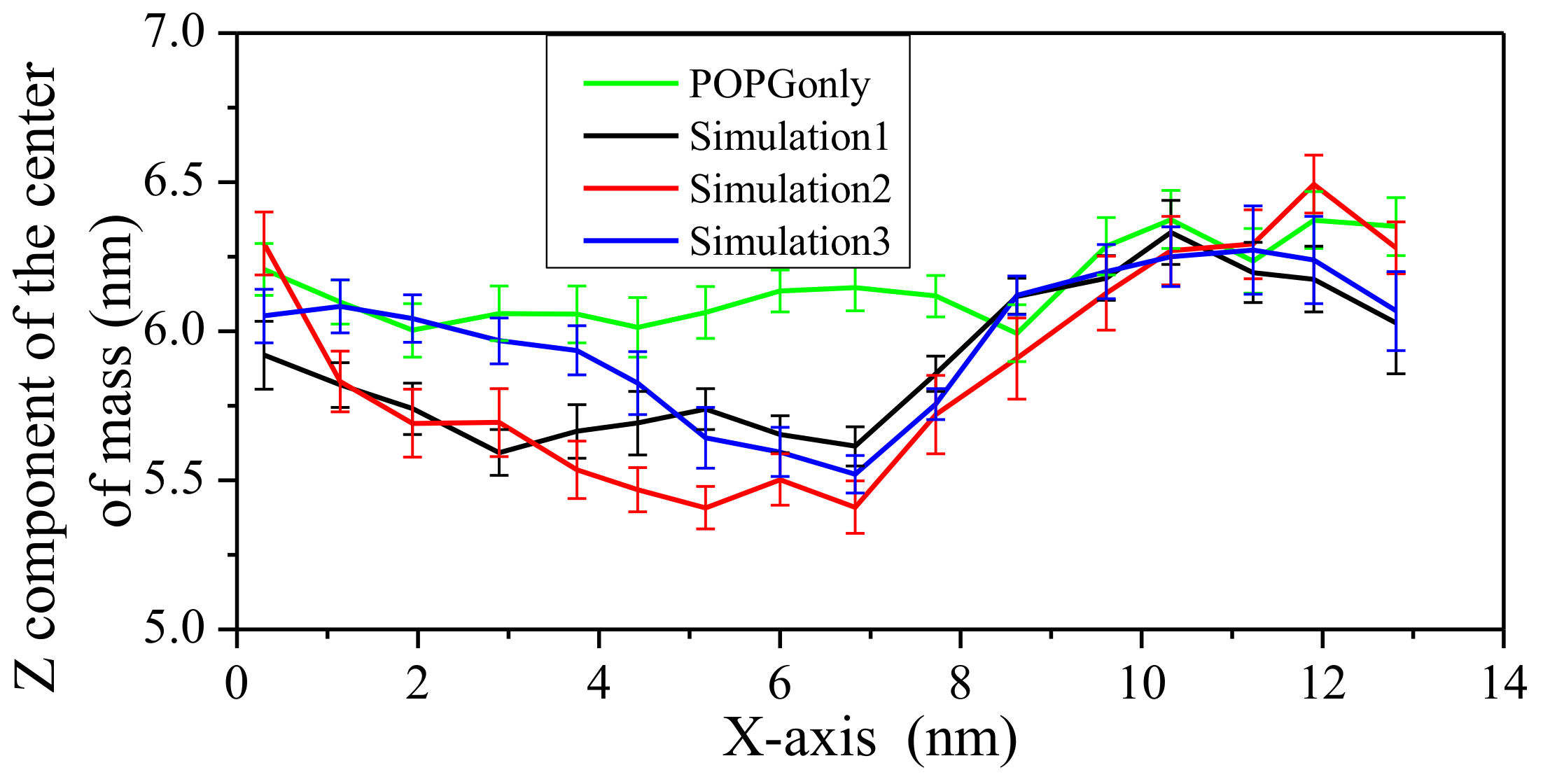
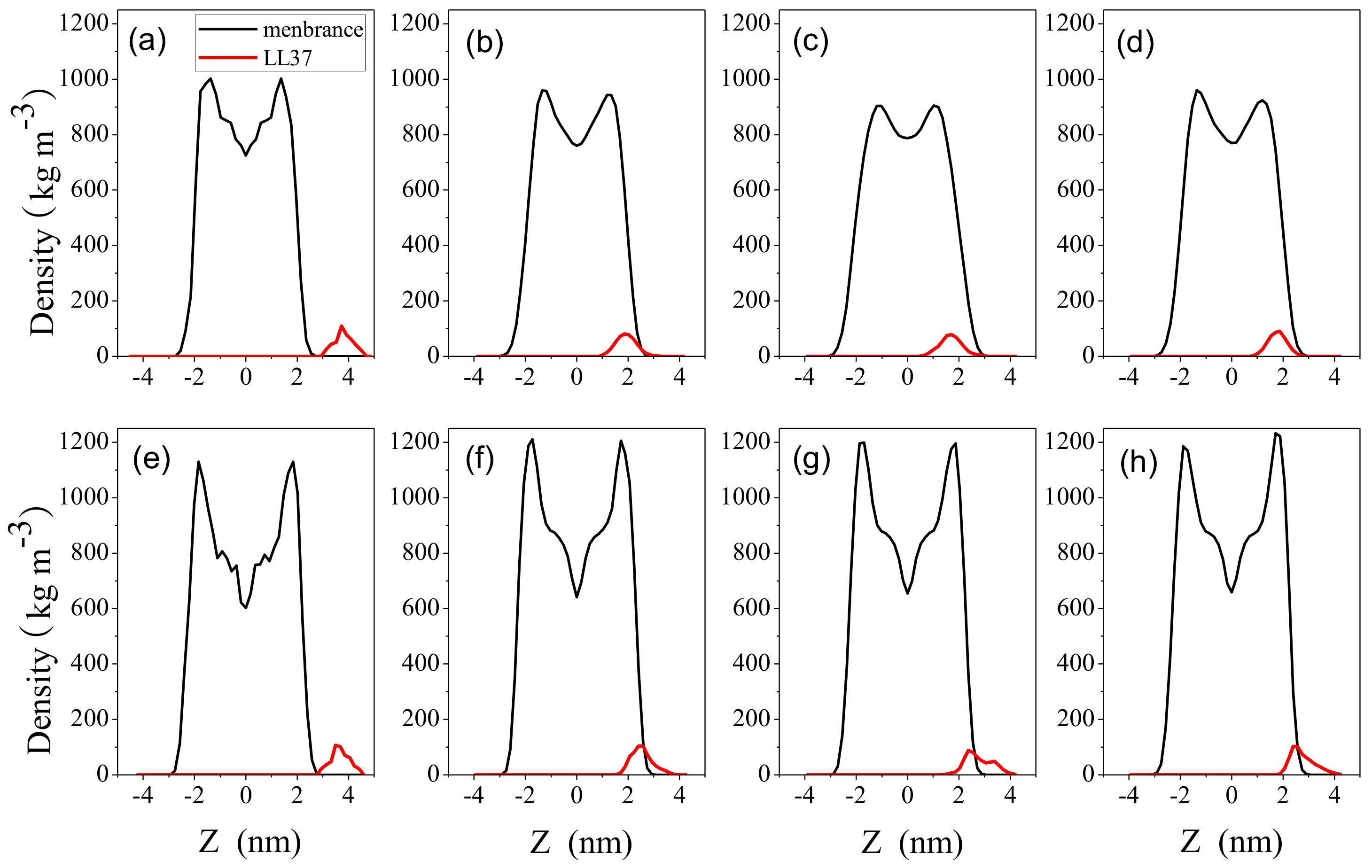
2.3. Impact on Membrane
3. Discussion
4. Materials and Methods
4.1. Modeling LL-37 and Membranes
4.2. Simulation Details
4.3. Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AMPs | antimicrobial peptides |
| NMR | nuclear magnetic resonance |
| MD | molecular dynamics |
| CG | coarse-grain |
| SASA | solvent accessible surface area |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol |
| POPS | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine |
| DPC | dodecylphosphocholine |
| DSC | differential scanning calorimetry |
| AFM | atomic force microscopy |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine |
| FTIR | Fourier-transform infrared |
References
- Wang, G. Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs. Pharmaceuticals 2013, 6, 728–758. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.; Mendoza, V.; Hancock, R.E. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 2002, 46, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert. Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Gwyer Findlay, E.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.; Wang, G.; Coffelt, S.B.; Betancourt, A.M.; Lee, C.W.; Fan, D.; Wu, K.; Yu, J.; Sung, J.J.; Cho, C.H. Emerging roles of the host defense peptide LL-37 in human cancer and its potential therapeutic applications. Int. J. Cancer 2010, 127, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Okumura, K.; Isogai, H.; Isogai, E. The Human Cathelicidin Antimicrobial Peptide LL-37 and Mimics are Potential Anticancer Drugs. Front. Oncol. 2015, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Kaplan, M.J. Little peptide, big effects: The role of LL-37 in inflammation and autoimmune disease. J. Immunol. 2013, 191, 4895–4901. [Google Scholar] [CrossRef] [PubMed]
- Noore, J.; Noore, A.; Li, B. Cationic antimicrobial peptide LL-37 is effective against both extra- and intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Duplantier, A.J.; van Hoek, M.L. The Human Cathelicidin Antimicrobial Peptide LL-37 as a Potential Treatment for Polymicrobial Infected Wounds. Front. Immunol. 2013, 4, 143. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1998, 273, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, F.; Verardi, R.; Shi, L.; Henzler-Wildman, K.A.; Ramamoorthy, A.; Veglia, G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Oglecka, K.; Sandgren, S.; Belting, M.; Esbjorner, E.K.; Norden, B.; Graslund, A. Dual functions of the human antimicrobial peptide LL-37-target membrane perturbation and host cell cargo delivery. Biochim. Biophys. Acta 2010, 1798, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, S.; Wittrup, A.; Cheng, F.; Jonsson, M.; Eklund, E.; Busch, S.; Belting, M. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J. Biol. Chem. 2004, 279, 17951–17956. [Google Scholar] [CrossRef] [PubMed]
- Seil, M.; Nagant, C.; Dehaye, J.P.; Vandenbranden, M.; Lensink, M.F. Spotlight on Human LL-37, an Immunomodulatory Peptide with Promising Cell-Penetrating Properties. Pharmaceuticals 2010, 3, 3435–3460. [Google Scholar] [CrossRef]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341 Pt 3, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef] [PubMed]
- Morgera, F.; Vaccari, L.; Antcheva, N.; Scaini, D.; Pacor, S.; Tossi, A. Primate cathelicidin orthologues display different structures and membrane interactions. Biochem. J. 2009, 417, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Neville, F.; Cahuzac, M.; Konovalov, O.; Ishitsuka, Y.; Lee, K.Y.; Kuzmenko, I.; Kale, G.M.; Gidalevitz, D. Lipid headgroup discrimination by antimicrobial peptide LL-37: Insight into mechanism of action. Biophys. J. 2006, 90, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Gable, J.E.; Schlamadinger, D.E.; Cogen, A.L.; Gallo, R.L.; Kim, J.E. Fluorescence and UV resonance Raman study of peptide-vesicle interactions of human cathelicidin LL-37 and its F6W and F17W mutants. Biochemistry 2009, 48, 11264–11272. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Sun, Y.; Qian, S.; Huang, H.W. Transmembrane pores formed by human antimicrobial peptide LL-37. Biophys. J. 2011, 100, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Soblosky, L.; Nguyen, K.; Geng, J.; Yu, X.; Ramamoorthy, A.; Chen, Z. Physiologically-relevant modes of membrane interactions by the human antimicrobial peptide, LL-37, revealed by SFG experiments. Sci. Rep. 2013, 3, 1854. [Google Scholar] [CrossRef] [PubMed]
- Sevcsik, E.; Pabst, G.; Richter, W.; Danner, S.; Amenitsch, H.; Lohner, K. Interaction of LL-37 with model membrane systems of different complexity: Influence of the lipid matrix. Biophys. J. 2008, 94, 4688–4699. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schlamadinger, D.E.; Kim, J.E.; McCammon, J.A. Comparative molecular dynamics simulations of the antimicrobial peptide CM15 in model lipid bilayers. Biochim. Biophys. Acta 2012, 1818, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.R.; Fujiwara, T. Membrane Mediated Antimicrobial and Antitumor Activity of Cathelicidin 6: Structural Insights from Molecular Dynamics Simulation on Multi-Microsecond Scale. PLoS ONE 2016, 11, e0158702. [Google Scholar] [CrossRef] [PubMed]
- Berglund, N.A.; Piggot, T.J.; Jefferies, D.; Sessions, R.B.; Bond, P.J.; Khalid, S. Interaction of the antimicrobial peptide polymyxin B1 with both membranes of E. coli: A molecular dynamics study. PLoS Comput. Biol. 2015, 11, e1004180. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, S.; Lakshminarayanan, R.; Bai, Y.; Pervushin, K.; Verma, C.; Beuerman, R.W. Molecular simulations suggest how a branched antimicrobial peptide perturbs a bacterial membrane and enhances permeability. Biochim. Biophys. Acta 2013, 1828, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P. Charged Antimicrobial Peptides Can Translocate across Membranes without Forming Channel-like Pores. Biophys. J. 2017, 113, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.J. Recent changes to RasMol, recombining the variants. Trends Biochem. Sci. 2000, 25, 453–455. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Manna, M.; Mukhopadhyay, C. Molecular dynamics simulations of the interactions of kinin peptides with an anionic POPG bilayer. Langmuir 2011, 27, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Tolokh, I.S.; Vivcharuk, V.; Tomberli, B.; Gray, C.G. Binding free energy and counterion release for adsorption of the antimicrobial peptide lactoferricin B on a POPG membrane. Phys. Rev. E 2009, 80, 031911. [Google Scholar] [CrossRef] [PubMed]
- Khandelia, H.; Langham, A.A.; Kaznessis, Y.N. Driving engineering of novel antimicrobial peptides from simulations of peptide–micelle interactions. Biochim. Biophys. Acta. 2006, 1758, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Ciornei, C.D.; Sigurdardottir, T.; Schmidtchen, A.; Bodelsson, M. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL-37. Antimicrob. Agents Chemother. 2005, 49, 2845–2850. [Google Scholar] [CrossRef] [PubMed]
- Kukol, A. Lipid Models for United-Atom Molecular Dynamics Simulations of Proteins. J. Chem. Theory. Comput. 2009, 5, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Von Deuster, C.I.; Knecht, V. Antimicrobial selectivity based on zwitterionic lipids and underlying balance of interactions. Biochim. Biophys. Acta 2012, 1818, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1981; p. 331. [Google Scholar]
- Berger, O.; Edholm, O.; Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Van der Waals Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy | Binding Energy | |
|---|---|---|---|---|---|---|
| LL-37/POPG | S1 | −2.1 ± 0.2 | −121.2 ± 2.1 | 16.1 ± 1.1 | −3.3 ± 0.1 | −110.5 ± 1.5 |
| S2 | −3.4 ± 0.2 | −116.0 ± 2.2 | 14.1 ± 1.2 | −3.3 ± 0.1 | −108.7 ± 1.8 | |
| S3 | −3.1 ± 0.1 | −118.8 ± 2.5 | 15.4 ± 1.4 | −3.3 ± 0.1 | −109.8 ± 1.5 | |
| LL-37/POPC | S1 | −2.7 ± 0.2 | −3.6 ± 0.5 | 6.7 ± 0.9 | 0.5 ± 0.1 | 1.0 ± 0.8 |
| S2 | −1.7 ± 0.4 | −3.8 ± 1.1 | 6.1 ± 1.4 | 0.6 ± 0.1 | 1.3 ± 0.7 | |
| S3 | −1.3 ± 0.5 | −1.0 ± 0.6 | 2.7 ± 1.15 | 0.7 ± 0.1 | 1.1 ± 0.4 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Cao, Z.; Bian, Y.; Hu, G.; Wang, J.; Zhou, Y. Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers. Int. J. Mol. Sci. 2018, 19, 1186. https://doi.org/10.3390/ijms19041186
Zhao L, Cao Z, Bian Y, Hu G, Wang J, Zhou Y. Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers. International Journal of Molecular Sciences. 2018; 19(4):1186. https://doi.org/10.3390/ijms19041186
Chicago/Turabian StyleZhao, Liling, Zanxia Cao, Yunqiang Bian, Guodong Hu, Jihua Wang, and Yaoqi Zhou. 2018. "Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers" International Journal of Molecular Sciences 19, no. 4: 1186. https://doi.org/10.3390/ijms19041186
APA StyleZhao, L., Cao, Z., Bian, Y., Hu, G., Wang, J., & Zhou, Y. (2018). Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers. International Journal of Molecular Sciences, 19(4), 1186. https://doi.org/10.3390/ijms19041186