β-Nicotinamide Adenine Dinucleotide (β-NAD) Inhibits ATP-Dependent IL-1β Release from Human Monocytic Cells

 ,
, 
Abstract
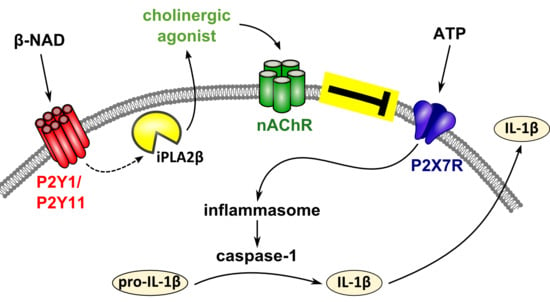
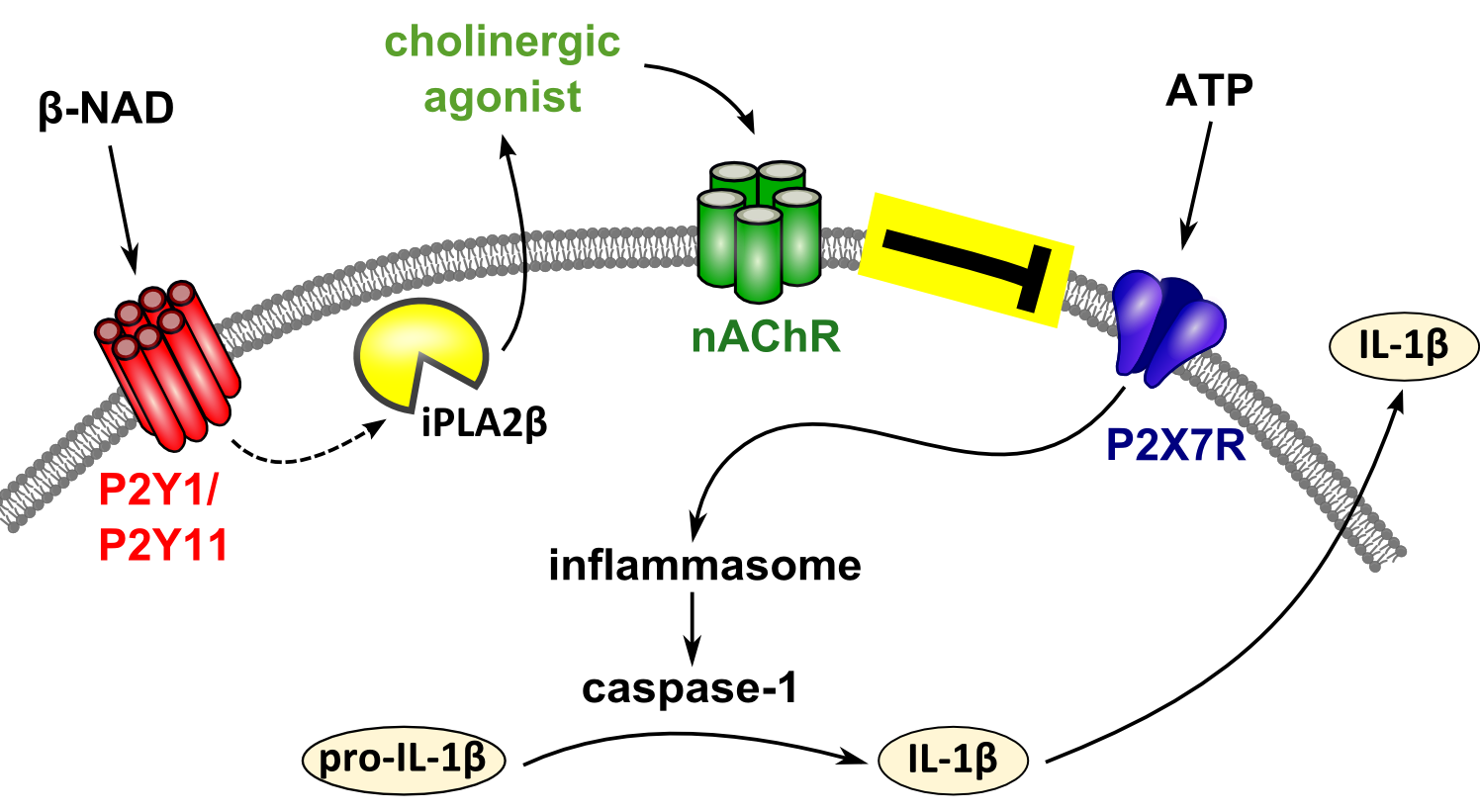{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
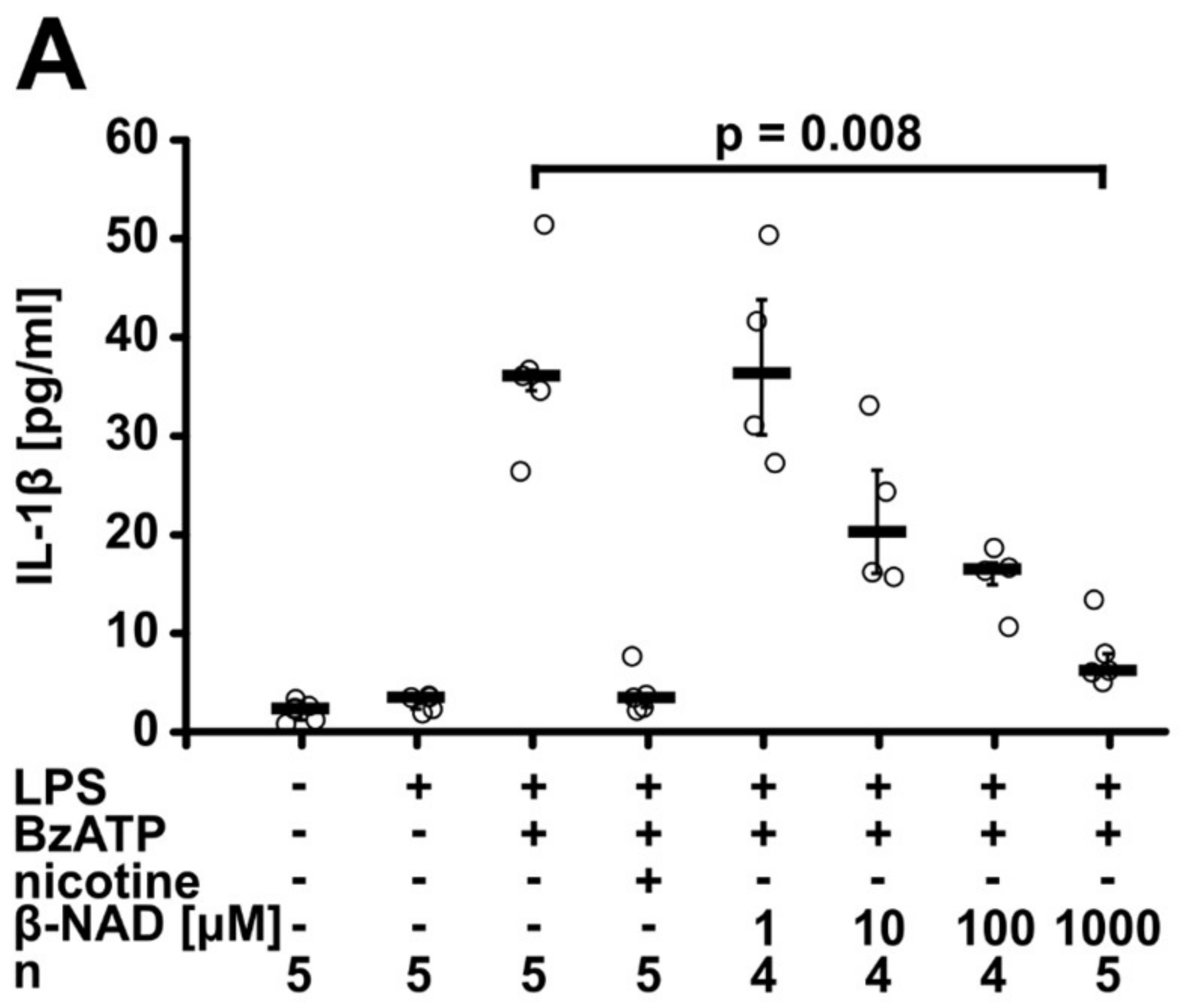
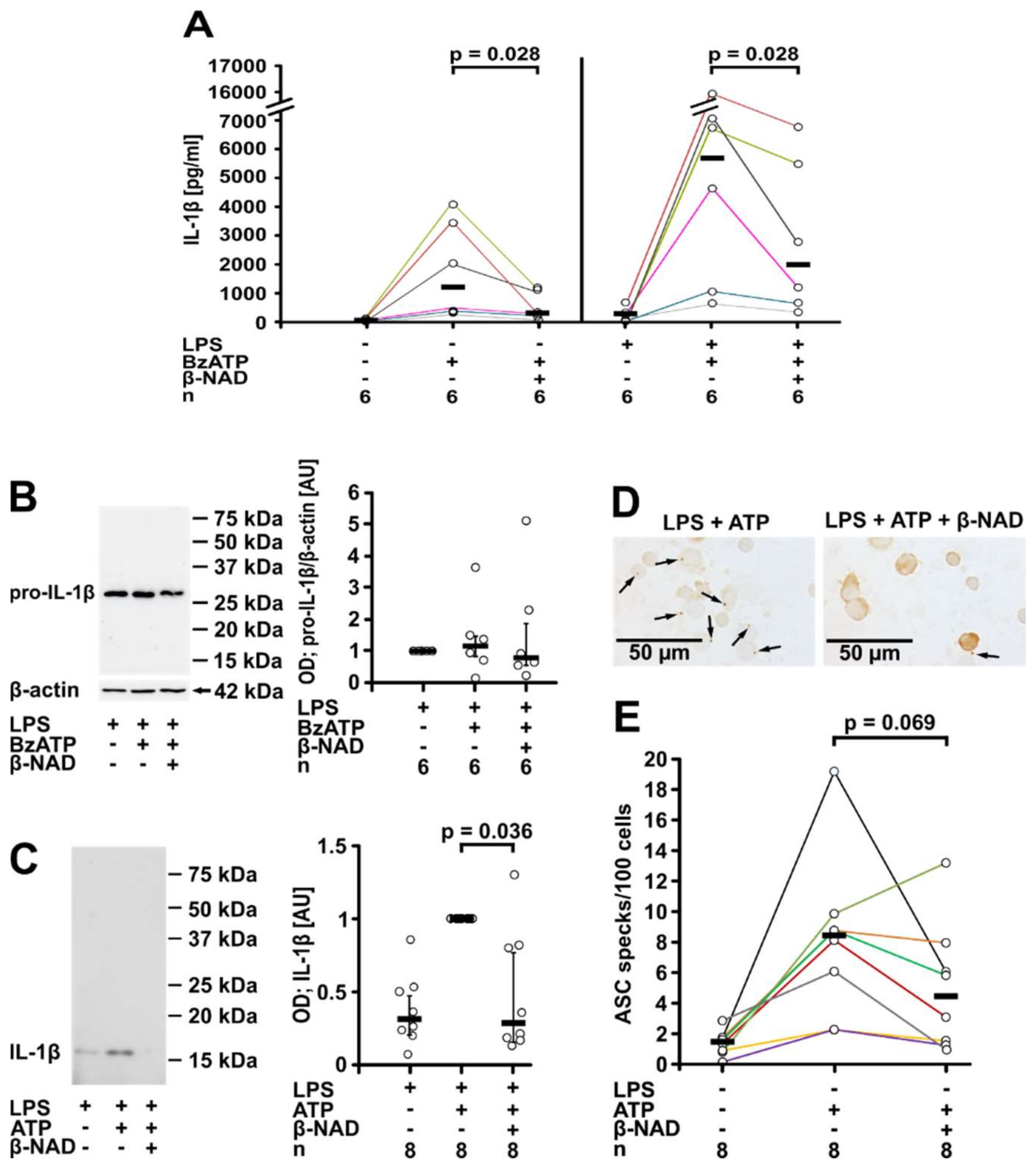
2.1. Inhibition of ATP-Mediated Release of IL-1β
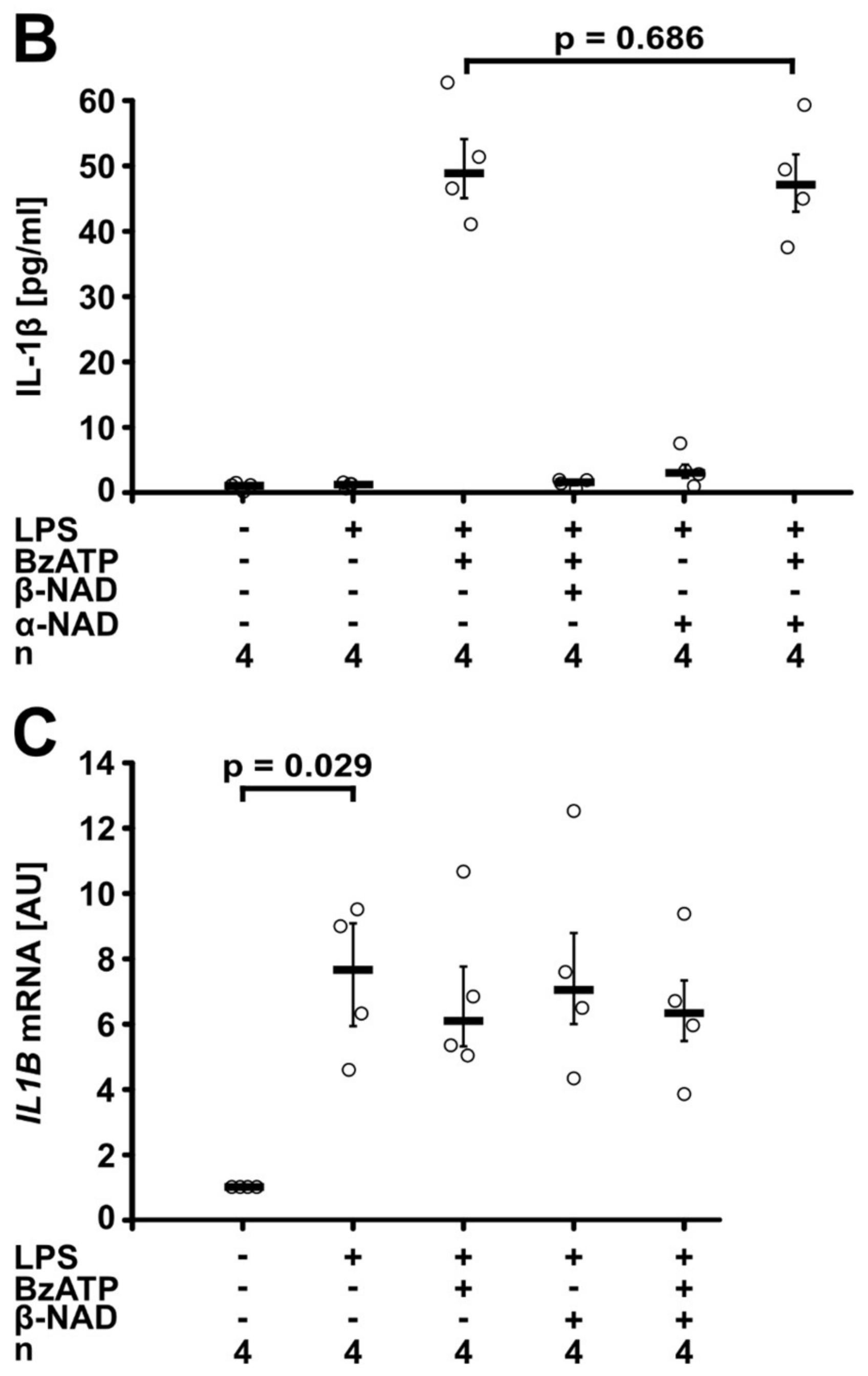
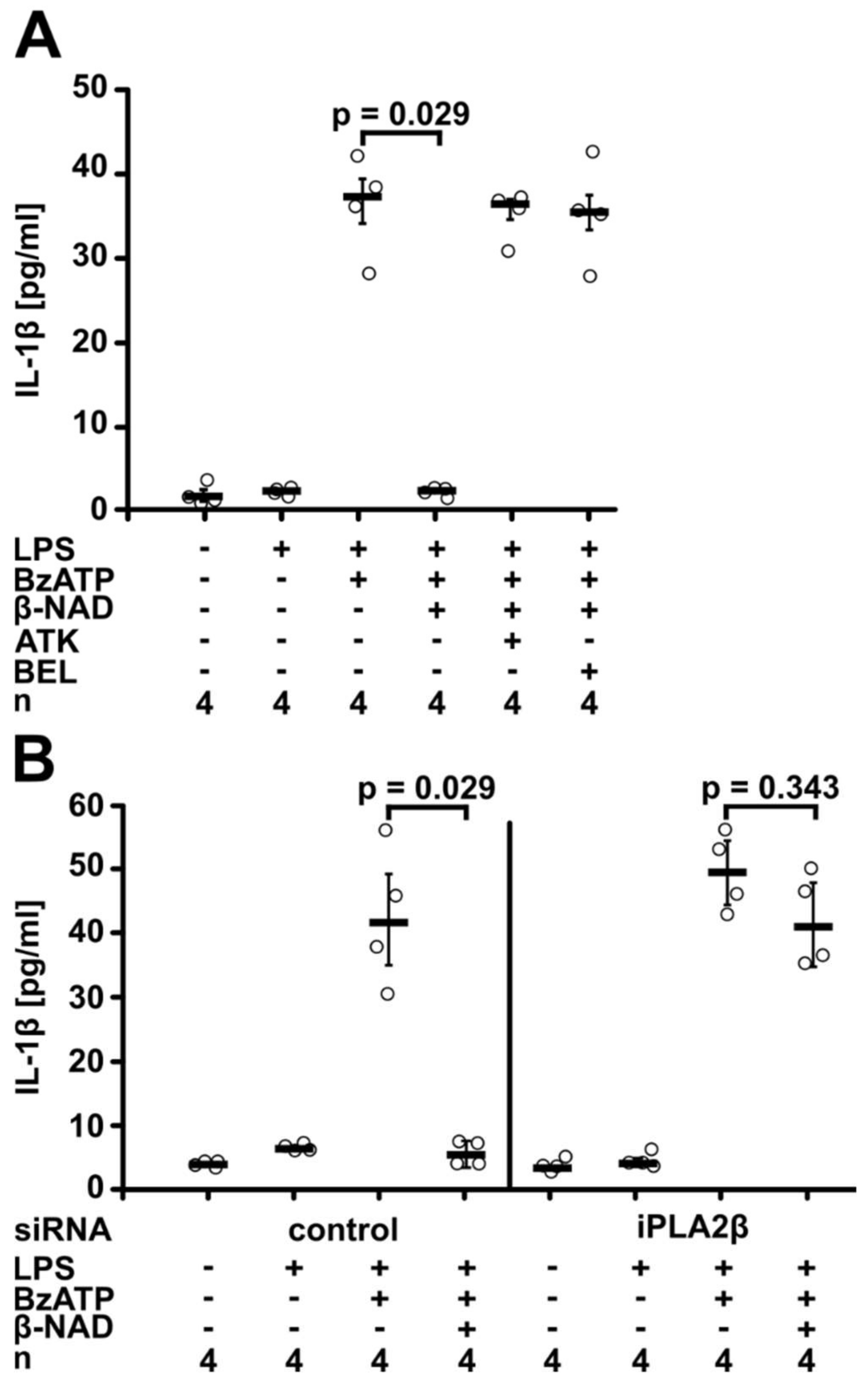
2.2. Involvement of Purinergic P2Y Receptors
2.3. Involvement of iPLA2β
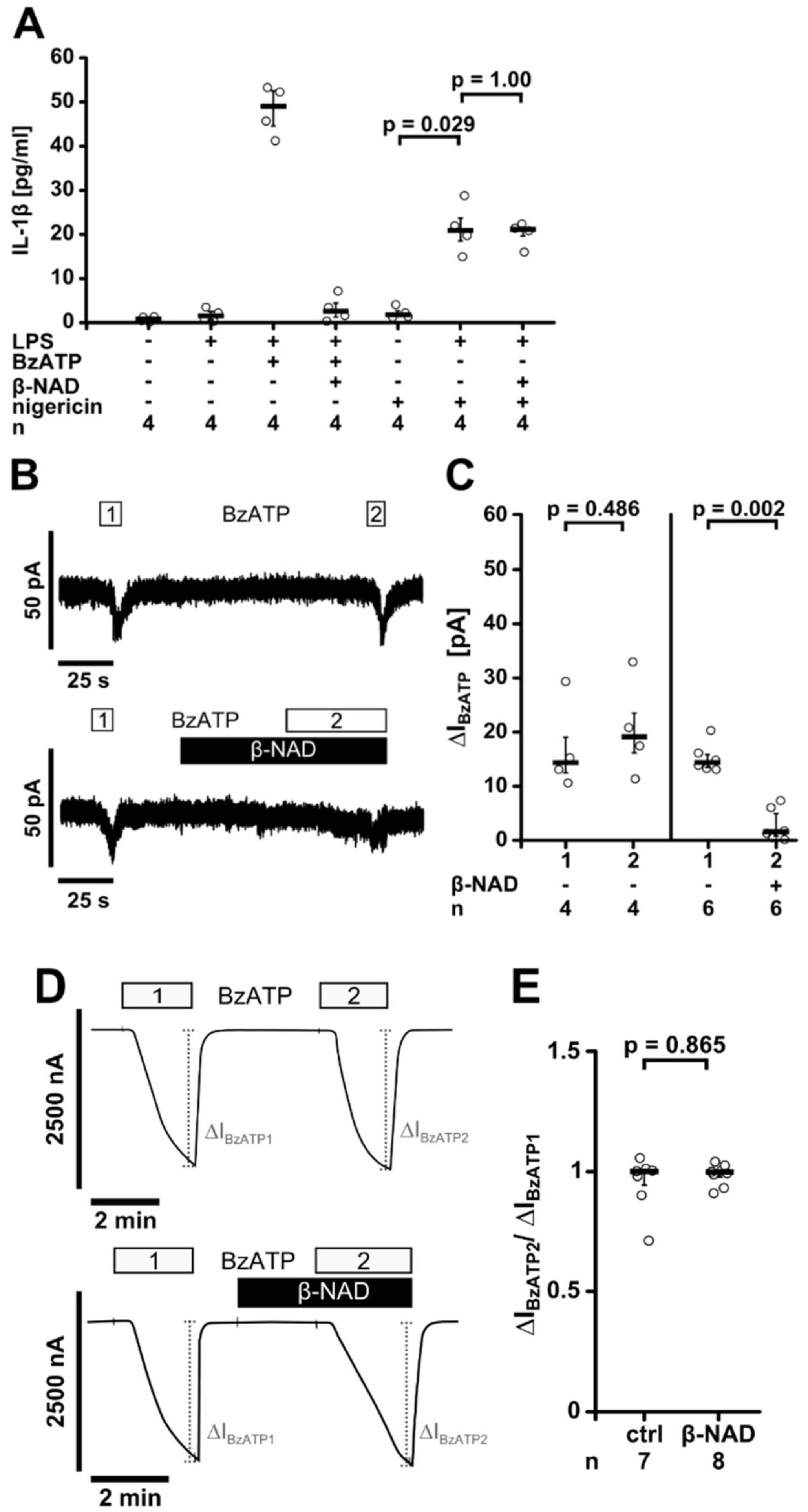
2.4. Release of a Soluble Factor
2.5. Involvement of nAChRs
2.6. Inhibition of P2X7 Receptor Function
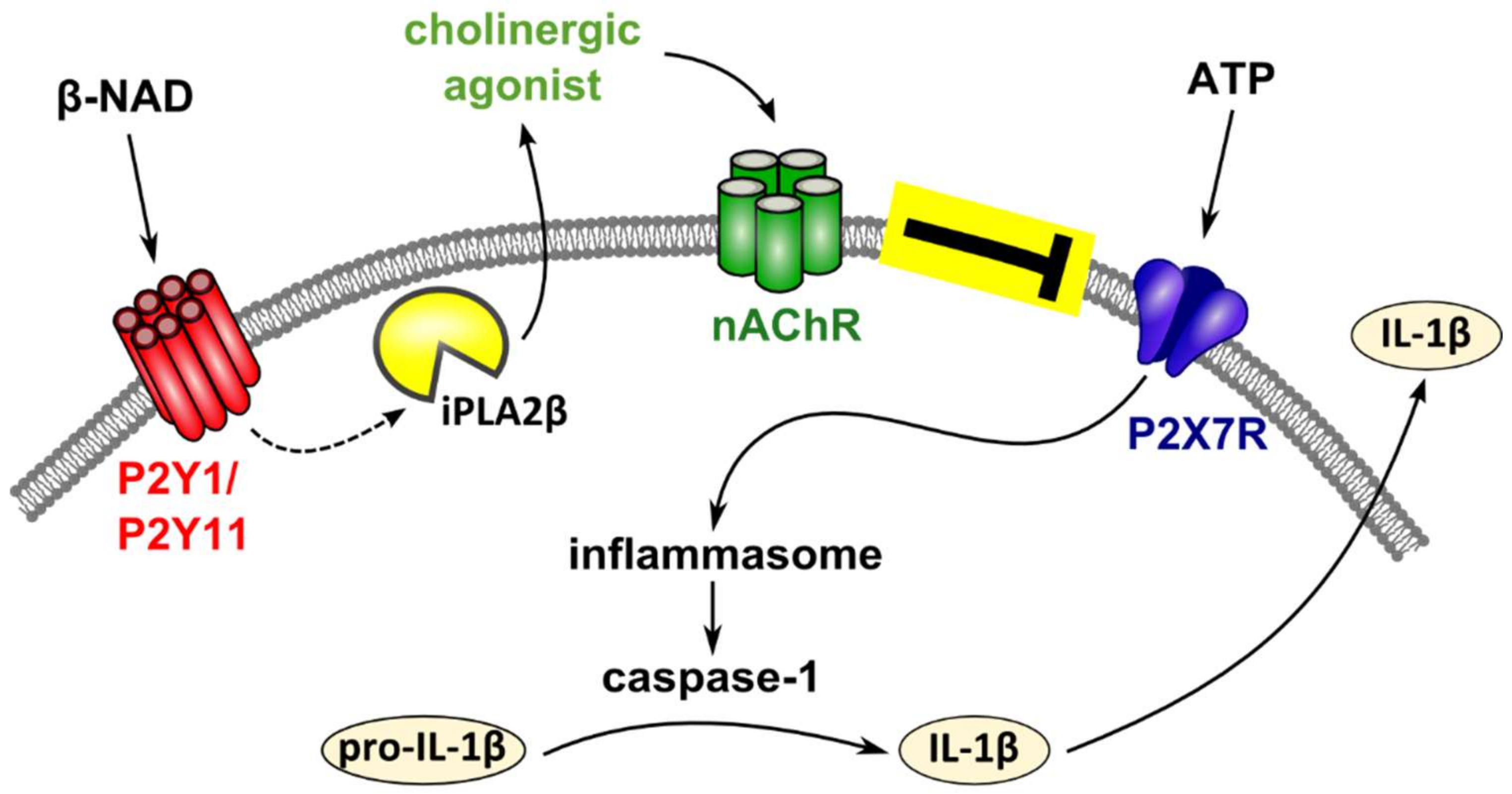
3. Discussion
4. Materials and Methods
4.1. U937 Cells
4.2. RNA Isolation and Real-Time RT-PCR
4.3. PBMCs
4.4. Immunocytochemistry
4.5. Gene Silencing
4.6. Western Blotting
4.7. Production and Use of Conditioned Medium
4.8. Patch-Clamp Experiments on U937 Cells
4.9. Heterologous Expression of Human P2X7 Receptor in Xenopus Laevis Oocytes and TEVC Measurements
4.10. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IL-1β | interleukin-1β |
| β-NAD | β-nicotinamide adenine dinucleotide |
| BzATP | 2′(3′)-O-(4-benzoyl-benzoyl)ATP trieethylammonium salt |
| nAChR | nicotinic acetylcholine receptor |
| PLA2 | phospholipase A2 |
| iPLA2β | Ca2+-independent phospholipase A2 |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| ACh | acetylcholine |
| SIRT3 | sirtuin-3 |
| SIRT2 | sirtuin-2 |
| ART2 | mono-ADP-ribosyltransferase A2 |
| LPS | lipopolysaccharide |
| LDH | lactate dehydrogenase |
| ATK | arachidonyl trifluoromethyl ketone |
| BEL | bromoenol lactone |
References
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.; Thiemermann, C.; Brohi, K. Trauma alarmins as activators of damage-induced inflammation. Br. J. Surg. 2012, 99 (Suppl. 1), 12–20. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome complexes: Emerging mechanisms and effector functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Hecker, A.; Küllmar, M.; Wilker, S.; Richter, K.; Zakrzewicz, A.; Atanasova, S.; Mathes, V.; Timm, T.; Klein, J.; Kaufmann, A.; et al. Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1β release. J. Immunol. 2015, 195, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Mathes, V.; Fronius, M.; Althaus, M.; Hecker, A.; Krasteva-Christ, G.; Padberg, W.; Hone, A.J.; McIntosh, J.M.; Zakrzewicz, A.; et al. Phosphocholine—An agonist of metabotropic but not of ionotropic functions of α9-containing nicotinic acetylcholine receptors. Sci. Rep. 2016, 6, 28660. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, A.; Richter, K.; Agné, A.; Wilker, S.; Siebers, K.; Fink, B.; Krasteva-Christ, G.; Althaus, M.; Padberg, W.; Hone, A.J.; et al. Canonical and novel non-canonical cholinergic agonists inhibit ATP-induced release of monocytic interleukin-1β via different combinations of nicotinic acetylcholine receptor subunits α7, α9 and α10. Front. Cell. Neurosci. 2017, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozakim, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Traba, J.; Kwarteng-Siaw, M.; Okoli, T.C.; Li, J.; Huffstutler, R.D.; Bray, A.; Waclawiw, M.A.; Han, K.; Pelletier, M.; Sauve, A.A.; et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Investig. 2015, 125, 4592–4600. [Google Scholar] [CrossRef] [PubMed]
- Harden, L.M.; Kent, S.; Pittman, Q.J.; Roth, J. Fever and sickness behavior: Friend or foe? Brain Behav. Immun. 2015, 50, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Hecker, A.; Mikulski, Z.; Lips, K.S.; Pfeil, U.; Zakrzewicz, A.; Wilker, S.; Hartmann, P.; Padberg, W.; Wessler, I.; Kummer, W.; et al. Pivotal advance: Upregulation of acetylcholine synthesis in intravascular transplant leukocytes during rejection of rat renal allografts. J. Leukoc. Biol. 2009, 86, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, A.; Kulikova, V.; Ziegler, M. The human NAD metabolome: Functions, metabolism and compartmentalization. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Moreschi, I.; Guida, L.; Usai, C.; Zocchi, E.; De Flora, A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem. J. 2006, 393, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Moreschi, I.; Bruzzone, S.; Nicholas, R.A.; Fruscione, F.; Sturla, L.; Benvenuto, F.; Usai, C.; Meis, S.; Kassack, M.U.; Zocchi, E.; De Flora, A. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J. Biol. Chem. 2006, 281, 31419–31429. [Google Scholar] [CrossRef] [PubMed]
- Adriouch, S.; Hubert, S.; Pechberty, S.; Koch-Nolte, F.; Haag, F.; Seman, M. NAD+ released during inflammation participates in T cell homeostasis by inducing ART2-mediated death of naive T cells in vivo. J. Immunol. 2007, 179, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Adriouch, S.; Bannas, P.; Schwarz, N.; Fliegert, R.; Guse, A.H.; Seman, M.; Haag, F.; Koch-Nolte, F. ADP-ribosylation at R125 gates the P2X7 ion channel by presenting a covalent ligand to its nucleotide binding site. FASEB J. 2008, 22, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Hubert, S.; Rissiek, B.; Klages, K.; Huehn, J.; Sparwasser, T.; Haag, F.; Koch-Nolte, F.; Boyer, O.; Seman, M.; Adriouch, S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J. Exp. Med. 2010, 207, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Pliyev, B.K.; Ivanova, A.V.; Savchenko, V.G. Extracellular NAD+ inhibits human neutrophil apoptosis. Apoptosis 2014, 19, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Rissiek, B.; Haag, F.; Boyer, O.; Koch-Nolte, F.; Adriouch, S. P2X7 on mouse T cells: One channel, many functions. Front. Immunol. 2015, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.; Adriouch, S.; Braasch, F.; Koestner, W.; Leiter, E.H.; Seman, M.; Lund, F.E.; Oppenheimer, N.; Haag, F.; Koch-Nolte, F. CD38 controls ADP-ribosyltransferase-2-catalyzed ADP-ribosylation of T cell surface proteins. J. Immunol. 2005, 174, 3298–3305. [Google Scholar] [CrossRef] [PubMed]
- Scheuplein, F.; Schwarz, N.; Adriouch, S.; Krebs, C.; Bannas, P.; Rissiek, B.; Seman, M.; Haag, F.; Koch-Nolte, F. NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J. Immunol. 2009, 182, 2898–2908. [Google Scholar] [CrossRef] [PubMed]
- Mutafova-Yambolieva, V.N.; Hwang, S.J.; Hao, X.; Chen, H.; Zhu, M.X.; Wood, J.D.; Ward, S.M.; Sanders, K.M. Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc. Natl. Acad. Sci. USA 2007, 104, 16359–16364. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Haag, F.; Koch-Nolte, F.; Kühl, M.; Lorenzen, S.; Thiele, H.G. Premature stop codons inactivate the RT6 genes of the human and chimpanzee species. J. Mol. Biol. 1994, 243, 537–546. [Google Scholar] [CrossRef] [PubMed]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [PubMed]
- Backhaus, S.; Zakrzewicz, A.; Richter, K.; Damm, J.; Wilker, S.; Fuchs-Moll, G.; Küllmar, M.; Hecker, A.; Manzini, I.; Ruppert, C.; et al. Surfactant inhibits ATP-induced release of interleukin-1β via nicotinic acetylcholine receptors. J. Lipid Res. 2017, 58, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L.; Adams, M.; Ravi, R.G.; Jacobson, K.A.; Harden, T.K. 2-Chloro N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate is a selective high affinity P2Y1 receptor antagonist. Br. J. Pharmacol. 2002, 135, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Dreisig, K.; Kornum, B.R. A critical look at the function of the P2Y11 receptor. Purinergic Signal. 2016, 12, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Bruner, G.; Murphy, S. Purinergic P2Y receptors on astrocytes are directly coupled to phospholipase A2. Glia 1993, 7, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wen, M.; Hayashi, J. Characterization of ATP receptor responsible for the activation of phospholipase A2 and stimulation of prostaglandin E2 production in thymic epithelial cells. Biochem. J. 1995, 308, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Chen, C.C. ATP-induced arachidonic acid release in cultured astrocytes is mediated by Gi protein coupled P2Y1 and P2Y2 receptors. Glia 1998, 22, 360–370. [Google Scholar] [CrossRef]
- Street, I.P.; Lin, H.K.; Laliberté, F.; Ghomashchi, F.; Wang, Z.; Perrier, H.; Tremblay, N.M.; Huang, Z.; Weech, P.K.; Gelb, M.H. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry 1993, 32, 5935–5940. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, E.J.; Conde-Frieboes, K.; Dennis, E.A. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J. Biol. Chem. 1995, 270, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Amati, A.L.; Zakrzewicz, A.; Siebers, R.; Wilker, S.; Heldmann, S.; Zakrzewicz, D.; Hecker, A.; McIntosh, J.M.; Padberg, W.; Grau, V. Chemokines (CCL3, CCL4, and CCL5) inhibit ATP-induced release of IL-1β by monocytic cells. Mediat. Inflamm. 2017, 2017, 1434872. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.R.; Zwart, R.; Sher, E.; Millar, N.S. Pharmacological properties of 9α10α nicotinic acetylcholine receptors revealed by heterologous expression of subunit chimeras. Mol. Pharmacol. 2004, 65, 453–460. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 2009, 78, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, D.; Shelukhina, I.; Vulfius, C.; Makarieva, T.; Stonik, V.; Zhmak, M.; Ivanov, I.; Kasheverov, I.; Utkin, Y.; Tsetlin, V. Natural compounds interacting with nicotinic acetylcholine receptors: From low-molecular weight ones to peptides and proteins. Toxins 2015, 7, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, synthesis, and structure activity of a highly selective α7 nicotinic acetylcholine receptor antagonist. Biochemistry 2007, 46, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Romero, H.K.; Christensen, S.B.; DiCesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef] [PubMed]
- Moser, N.; Mechawar, N.; Jones, I.; Gochberg-Sarver, A.; Orr-Urtreger, A.; Plomann, M.; Salas, R.; Molles, B.; Marubio, L.; Roth, U.; et al. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J. Neurochem. 2007, 102, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Rommel, F.R.; Raghavan, B.; Paddenberg, R.; Kummer, W.; Tumala, S.; Lochnit, G.; Gieler, U.; Peters, E.M. Suitability of nicotinic acetylcholine receptor α7 and muscarinic acetylcholine receptor 3 antibodies for immune detection: Evaluation in murine skin. J. Histochem. Cytochem. 2015, 63, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; Macpherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Gerth, A.; Nieber, K.; Oppenheimer, N.J.; Hauschildt, S. Extracellular NAD+ regulates intracellular free calcium concentration in human monocytes. Biochem. J. 2004, 382, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, N.S.; Zemskov, E.A.; Gonzales, J.; Gorshkov, B.A.; Sridhar, S.; Chakraborty, T.; Lucas, R.; Verin, A.D. Extracellular β-nicotinamide adenine dinucleotide (β-NAD) promotes the endothelial cell barrier integrity via PKA- and EPAC1/Rac1-dependent actin cytoskeleton rearrangement. J. Cell. Physiol. 2010, 223, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, N.S.; Gonzales, J.; Fulzele, S.; Kim, K.M.; Lucas, R.; Verin, A.D. β-Nicotinamide adenine dinucleotide attenuates lipopolysaccharide-induced inflammatory effects in a murine model of acute lung injury. Exp. Lung Res. 2012, 38, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Grahnert, A.; Abdelrahman, A.; Müller, C.E.; Hauschildt, S. Extracellular NAD+ induces a rise in [Ca2+]i in activated human monocytes via engagement of P2Y(1) and P2Y(11) receptors. Cell Calcium 2009, 46, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, L.; Gorini, S.; Rosano, G.; la Sala, A. Immunoregulation through extracellular nucleotides. Blood 2012, 120, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Ecke, D.; Hanck, T.; Tulapurkar, M.E.; Schäfer, R.; Kassack, M.; Stricker, R.; Reiser, G. Hetero-oligomerization of the P2Y11 receptor with the P2Y1 receptor controls the internalization and ligand selectivity of the P2Y11 receptor. Biochem. J. 2008, 409, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kudo, I.; Murakami, M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002, 68–69, 3–58. [Google Scholar] [CrossRef]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 2011, 63, 641–683. [Google Scholar] [CrossRef] [PubMed]
- Innocent, N.; Livingstone, P.D.; Hone, A.; Kimura, A.; Young, T.; Whiteaker, P.; McIntosh, J.M.; Wonnacott, S. Alpha-conotoxin Arenatus IB[V11L,V16D] [corrected] is a potent and selective antagonist at rat and human native alpha7 nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2008, 327, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Klapperstück, M.; Büttner, C.; Schmalzing, G.; Markwardt, F. Functional evidence of distinct ATP activation sites at the human P2X(7) receptor. J. Physiol. 2001, 534, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Stolz, M.; Klapperstück, M.; Kendzierski, T.; Detro-Dassen, S.; Panning, A.; Schmalzing, G.; Markwardt, F. Homodimeric anoctamin-1, but not homodimeric anoctamin-6, is activated by calcium increases mediated by the P2Y1 and P2X7 receptors. Pflugers Arch. 2015, 467, 2121–2140. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiller, S.D.; Heldmann, S.; Richter, K.; Jurastow, I.; Küllmar, M.; Hecker, A.; Wilker, S.; Fuchs-Moll, G.; Manzini, I.; Schmalzing, G.; et al. β-Nicotinamide Adenine Dinucleotide (β-NAD) Inhibits ATP-Dependent IL-1β Release from Human Monocytic Cells. Int. J. Mol. Sci. 2018, 19, 1126. https://doi.org/10.3390/ijms19041126
Hiller SD, Heldmann S, Richter K, Jurastow I, Küllmar M, Hecker A, Wilker S, Fuchs-Moll G, Manzini I, Schmalzing G, et al. β-Nicotinamide Adenine Dinucleotide (β-NAD) Inhibits ATP-Dependent IL-1β Release from Human Monocytic Cells. International Journal of Molecular Sciences. 2018; 19(4):1126. https://doi.org/10.3390/ijms19041126
Chicago/Turabian StyleHiller, Sebastian Daniel, Sarah Heldmann, Katrin Richter, Innokentij Jurastow, Mira Küllmar, Andreas Hecker, Sigrid Wilker, Gabriele Fuchs-Moll, Ivan Manzini, Günther Schmalzing, and et al. 2018. "β-Nicotinamide Adenine Dinucleotide (β-NAD) Inhibits ATP-Dependent IL-1β Release from Human Monocytic Cells" International Journal of Molecular Sciences 19, no. 4: 1126. https://doi.org/10.3390/ijms19041126
APA StyleHiller, S. D., Heldmann, S., Richter, K., Jurastow, I., Küllmar, M., Hecker, A., Wilker, S., Fuchs-Moll, G., Manzini, I., Schmalzing, G., Kummer, W., Padberg, W., McIntosh, J. M., Damm, J., Zakrzewicz, A., & Grau, V. (2018). β-Nicotinamide Adenine Dinucleotide (β-NAD) Inhibits ATP-Dependent IL-1β Release from Human Monocytic Cells. International Journal of Molecular Sciences, 19(4), 1126. https://doi.org/10.3390/ijms19041126