Potential Interplay between Hyperosmolarity and Inflammation on Retinal Pigmented Epithelium in Pathogenesis of Diabetic Retinopathy



Abstract
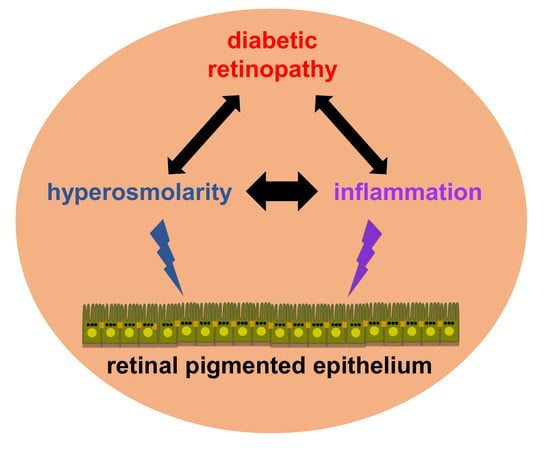
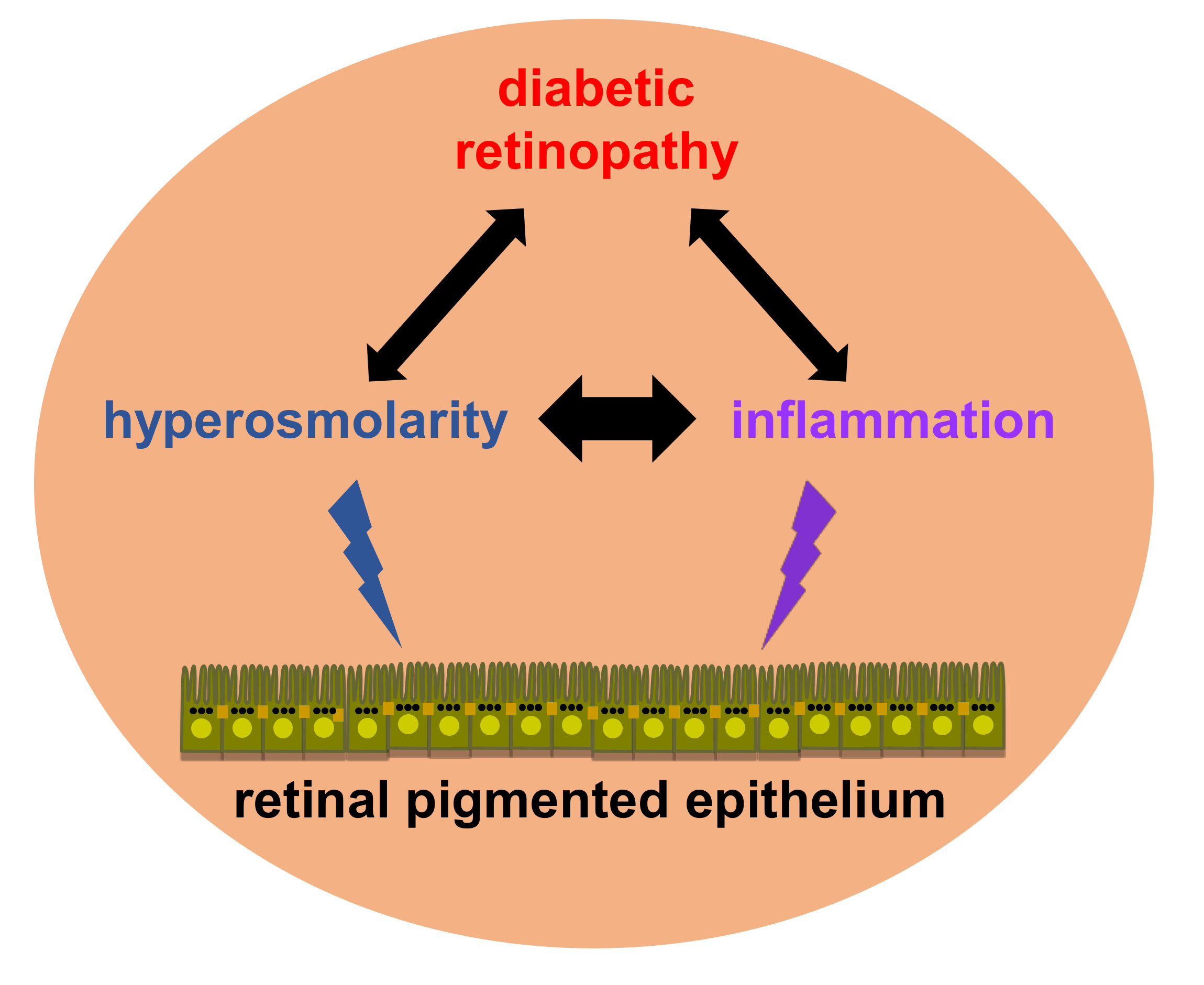
1. Introduction
2. What Are the General Characteristics and Functions of RPE Cells?
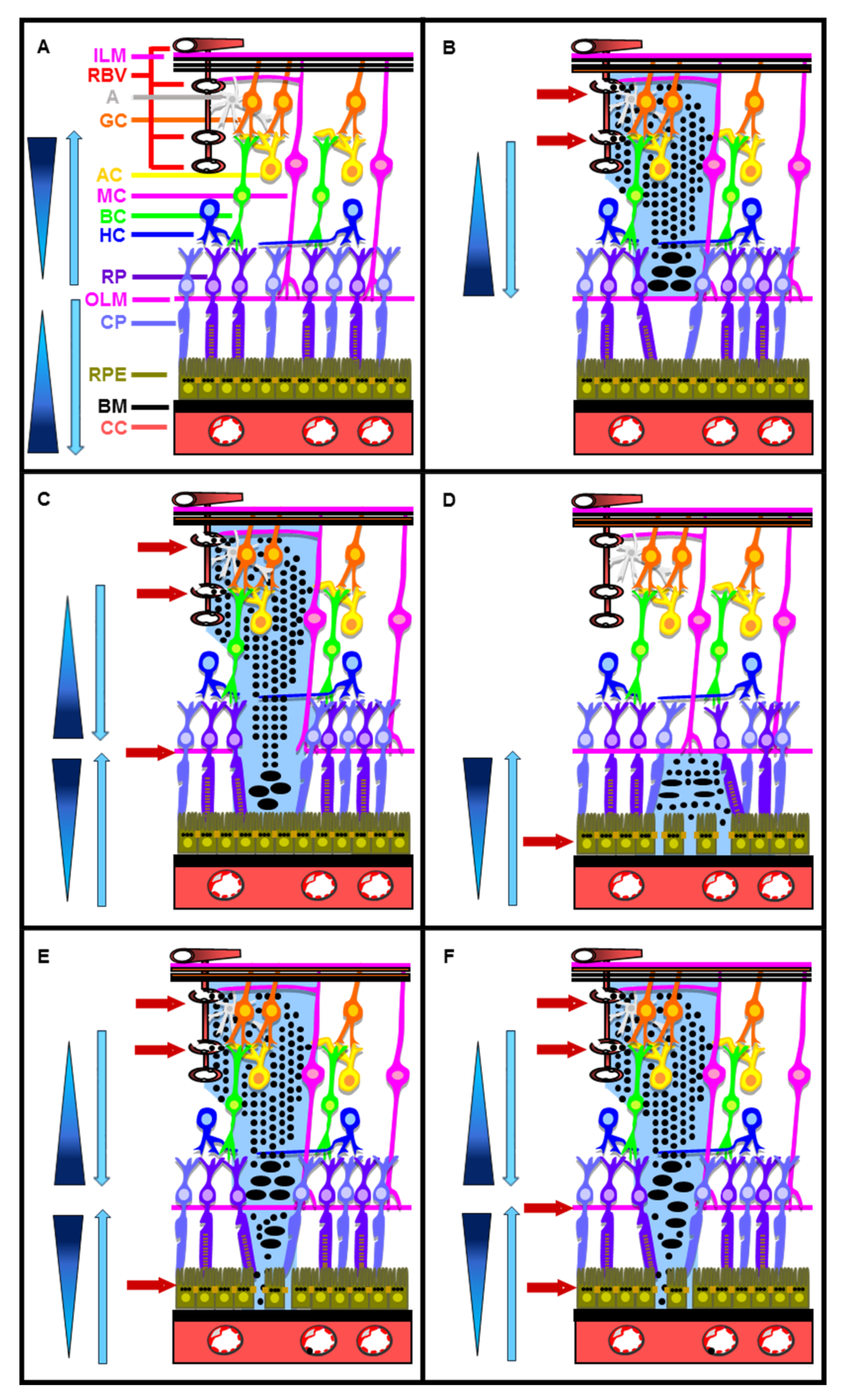
3. How Is Water Present in the Subretinal Space, Eliminated by the RPE Cells?
4. What Is the Role of Inflammation in BRB Rupture Occurring during DR?
5. What Are the Effects of HOS on Innate and Adaptive Immune Responses?
6. How Can RPE Cells Be Subjected to HOS during DR?
7. What Are the Consequences of HOS on the RPE?
8. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AQP | Aquaporin |
| AMD | Age-related macular degeneration |
| BRB | Blood retinal barrier |
| DR | Diabetic retinopathy |
| HOS | Hyperosmolar stress |
| HSD | High salt diet |
| iBRB | Inner blood retinal barrier |
| ILM | Inner limiting membrane |
| NFAT5 | Nuclear factor of activated T-cells 5 |
| TonEBP | Tonicity-responsive binding protein |
| oBRB | Outer blood retinal barrier |
| OLM | Outer limiting membrane |
| RPE | Retinal pigmented epithelium |
References
- Kolb, H. Simple Anatomy of the Retina. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Hosoya, K.; Tachikawa, M. The inner blood-retinal barrier: Molecular structure and transport biology. Adv. Exp. Med. Biol. 2012, 763, 85–104. [Google Scholar] [PubMed]
- Willermain, F.; Libert, S.; Motulsky, E.; Salik, D.; Caspers, L.; Perret, J.; Delporte, C. Origins and consequences of hyperosmolar stress in retinal pigmented epithelial cells. Front. Physiol. 2014, 5, 199. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Giovannelli, G.; Confalone, P.; Renna, F.V.; Geng, Y.-J.; De Caterina, R. High glucose-induced hyperosmolarity contributes to COX-2 expression and angiogenesis: Implications for diabetic retinopathy. Cardiovasc. Diabetol. 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Hollborn, M.; Kohen, L.; Wiedemann, P. Intake of dietary salt and drinking water: Implications for the development of age-related macular degeneration. Mol. Vis. 2016, 22, 1437–1454. [Google Scholar] [PubMed]
- Qin, Y.; Xu, G.; Fan, J.; Witt, R.E.; Da, C. High-salt loading exacerbates increased retinal content of aquaporins AQP1 and AQP4 in rats with diabetic retinopathy. Exp. Eye Res. 2009, 89, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, C.; Yoshimura, Y.; Kamada, C.; Tanaka, S.; Tanaka, S.; Hanyu, O.; Araki, A.; Ito, H.; Tanaka, A.; Ohashi, Y.; et al. Japan Diabetes Complications Study Group Dietary sodium intake and incidence of diabetes complications in Japanese patients with type 2 diabetes: Analysis of the Japan Diabetes Complications Study (JDCS). J. Clin. Endocrinol. Metab. 2014, 99, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Baldo, M.P.; Rodrigues, S.L.; Mill, J.G. High salt intake as a multifaceted cardiovascular disease: New support from cellular and molecular evidence. Heart Fail. Rev. 2015, 20, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Aaron, K.J.; Sanders, P.W. Role of dietary salt and potassium intake in cardiovascular health and disease: A review of the evidence. Mayo Clin. Proc. 2013, 88, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.M.G.; Wong, T.Y. Is age-related macular degeneration a manifestation of systemic disease? New prospects for early intervention and treatment. J. Intern. Med. 2014, 276, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Kostraba, J.N.; Klein, R.; Dorman, J.S.; Becker, D.J.; Drash, A.L.; Maser, R.E.; Orchard, T.J. The epidemiology of diabetes complications study. IV. Correlates of diabetic background and proliferative retinopathy. Am. J. Epidemiol. 1991, 133, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Dow, C.; Mancini, F.; Rajaobelina, K.; Boutron-Ruault, M.-C.; Balkau, B.; Bonnet, F.; Fagherazzi, G. Diet and risk of diabetic retinopathy: A systematic review. Eur. J. Epidemiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hucke, S.; Eschborn, M.; Liebmann, M.; Herold, M.; Freise, N.; Engbers, A.; Ehling, P.; Meuth, S.G.; Roth, J.; Kuhlmann, T.; et al. Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J. Autoimmun. 2016, 67, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Manzel, A.; Muller, D.N.; Hafler, D.A.; Erdman, S.E.; Linker, R.A.; Kleinewietfeld, M. Role of “Western diet” in inflammatory autoimmune diseases. Curr. Allergy Asthma Rep. 2014, 14, 404. [Google Scholar] [CrossRef] [PubMed]
- Krementsov, D.N.; Case, L.K.; Hickey, W.F.; Teuscher, C. Exacerbation of autoimmune neuroinflammation by dietary sodium is genetically controlled and sex specific. FASEB J. 2015, 29, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Hucke, S.; Wiendl, H.; Klotz, L. Implications of dietary salt intake for multiple sclerosis pathogenesis. Mult. Scler. Houndmills Basingstoke Engl. 2016, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Subramaniyam, G. Molecular underpinnings of Th17 immune-regulation and their implications in autoimmune diabetes. Cytokine 2015, 71, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Sato, T.; Sakurai, Y.; Taguchi, M.; Harimoto, K.; Karasawa, Y.; Ito, M. Association between aqueous humor and vitreous fluid levels of Th17 cell-related cytokines in patients with proliferative diabetic retinopathy. PLoS ONE 2017, 12, e0178230. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Wang, Y.; Zhu, G.; Gu, Y.; Mao, L.; Hong, M.; Li, Y.; Zheng, M. Imbalance of Th17/Treg cells in pathogenesis of patients with human leukocyte antigen B27 associated acute anterior uveitis. Sci. Rep. 2017, 7, 40414. [Google Scholar] [CrossRef] [PubMed]
- Guedes, M.C.E.; Borrego, L.M.; Proença, R.D. Roles of interleukin-17 in uveitis. Indian J. Ophthalmol. 2016, 64, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.S.; Steinberg, R.H. Passive ionic properties of frog retinal pigment epithelium. J. Membr. Biol. 1977, 36, 337–372. [Google Scholar] [CrossRef] [PubMed]
- Bok, D. The retinal pigment epithelium: A versatile partner in vision. J. Cell Sci. Suppl. 1993, 17, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D. The polarity of the retinal pigment epithelium. Traffic 2001, 2, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Lerche, W. Electron microscope studies on the differentiation of the pigmented epithelium and external granular cells (sensory cells) in the human eye. Zeitschrift fur Zellforschung und Mikroskopische Anatomie 1963, 58, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef] [PubMed]
- Detrick, B.; Hooks, J.J. Immune regulation in the retina. Immunol. Res. 2010, 47, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, S.; Karl, M.O.; Strauss, O. Ion channels in the RPE. Prog. Retin. Eye Res. 2007, 26, 263–301. [Google Scholar] [CrossRef] [PubMed]
- Agre, P. Aquaporin water channels (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2004, 43, 4278–4290. [Google Scholar] [CrossRef] [PubMed]
- Juuti-Uusitalo, K.; Delporte, C.; Grégoire, F.; Perret, J.; Huhtala, H.; Savolainen, V.; Nymark, S.; Hyttinen, J.; Uusitalo, H.; Willermain, F.; et al. Aquaporin expression and function in human pluripotent stem cell-derived retinal pigmented epithelial cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3510–3519. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Dukic-Stefanovic, S.; Pannicke, T.; Ulbricht, E.; Reichenbach, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Expression of aquaporins in the retina of diabetic rats. Curr. Eye Res. 2011, 36, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Rehak, M.; Iandiev, I.; Pannicke, T.; Ulbricht, E.; Reichenbach, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Transcriptional regulation of aquaporins in the ischemic rat retina: Upregulation of aquaporin-9. Curr. Eye Res. 2012, 37, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Ortak, H.; Cayli, S.; Ocaklı, S.; Söğüt, E.; Ekici, F.; Tas, U.; Demir, S. Age-related changes of aquaporin expression patterns in the postnatal rat retina. Acta Histochem. 2013, 115, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Köferl, P.; Hollborn, M.; Rehak, J.; Iandiev, I.; Dukic-Stefanovic, S.; Wiedemann, P.; Kohen, L.; Bringmann, A.; Rehak, M. Effects of arteriolar constriction on retinal gene expression and Müller cell responses in a rat model of branch retinal vein occlusion. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Stamer, W.D.; Bok, D.; Hu, J.; Jaffe, G.J.; McKay, B.S. Aquaporin-1 channels in human retinal pigment epithelium: Role in transepithelial water movement. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2803–2808. [Google Scholar] [CrossRef]
- Hamann, S.; Zeuthen, T.; La Cour, M.; Nagelhus, E.A.; Ottersen, O.P.; Agre, P.; Nielsen, S. Aquaporins in complex tissues: Distribution of aquaporins 1–5 in human and rat eye. Am. J. Physiol. 1998, 274, C1332–C1345. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.H.; Verkman, A.S. Aquaporins and CFTR in ocular epithelial fluid transport. J. Membr. Biol. 2006, 210, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.L.; Bek, T.; la Cour, M.; Prause, J.U.; Hamann, S.; Heegaard, S. Aquaporin-1 Expression in Retinal Pigment Epithelial Cells Overlying Retinal Drusen. Ophthalmic Res. 2016, 55, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Ulbricht, E.; Reichenbach, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Transcriptional regulation of aquaporin-3 in human retinal pigment epithelial cells. Mol. Biol. Rep. 2012, 39, 7949–7956. [Google Scholar] [CrossRef] [PubMed]
- Rehak, M.; Drechsler, F.; Köferl, P.; Hollborn, M.; Wiedemann, P.; Bringmann, A.; Kohen, L. Effects of intravitreal triamcinolone acetonide on retinal gene expression in a rat model of central retinal vein occlusion. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Vogler, S.; Reichenbach, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Regulation of the hyperosmotic induction of aquaporin 5 and VEGF in retinal pigment epithelial cells: Involvement of NFAT5. Mol. Vis. 2015, 21, 360–377. [Google Scholar] [PubMed]
- Tran, T.L.; Bek, T.; Holm, L.; la Cour, M.; Nielsen, S.; Prause, J.U.; Rojek, A.; Hamann, S.; Heegaard, S. Aquaporins 6–12 in the human eye. Acta Ophthalmol. 2013, 91, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian J. Ophthalmol. 2012, 60, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Vaz, J. Mechanisms of retinal fluid accumulation and blood-retinal barrier breakdown. Dev. Ophthalmol. 2017, 58, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Eshaq, R.S.; Aldalati, A.M.Z.; Alexander, J.S.; Harris, N.R. Diabetic retinopathy: Breaking the barrier. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2017, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.S.; Aiello, L.P.; Ferris, F.L.; Klein, R. Diabetic retinopathy. Diabetes Care 2004, 27, 2540–2553. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Kirste, G.; Schrader, W. Progressive proliferative diabetic retinopathy after transplantation of the pancreas. A case and a review of the topic. Acta Ophthalmol. 1994, 72, 743–751. [Google Scholar] [CrossRef]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.-P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Alhomida, A.S.; Ferrario, C.M.; Ahmad, S. Role of Tissue Renin-angiotensin system and the chymase/angiotensin-(1–12) axis in the pathogenesis of diabetic retinopathy. Curr. Med. Chem. 2017, 24, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Khullar, M.; Cheema, B.S.; Raut, S.K. Emerging evidence of epigenetic modifications in vascular complication of diabetes. Front. Endocrinol. 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Diabetic retinopathy, metabolic memory and epigenetic modifications. Vis. Res. 2017, 139, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Adamis, A.P.; Berman, A.J. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin. Immunopathol. 2008, 30, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.F.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic retinopathy: vascular and inflammatory disease. J. Diabetes Res. 2015, 2015, 582060. [Google Scholar] [CrossRef] [PubMed]
- Pusparajah, P.; Lee, L.-H.; Abdul Kadir, K. Molecular markers of diabetic retinopathy: Potential screening tool of the future? Front. Physiol. 2016, 7, 200. [Google Scholar] [CrossRef] [PubMed]
- Paccola, L.; Costa, R.A.; Folgosa, M.S.; Barbosa, J.C.; Scott, I.U.; Jorge, R. Intravitreal triamcinolone versus bevacizumab for treatment of refractory diabetic macular oedema (IBEME study). Br. J. Ophthalmol. 2008, 92, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Shimura, M.; Nakazawa, T.; Yasuda, K.; Shiono, T.; Iida, T.; Sakamoto, T.; Nishida, K. Comparative therapy evaluation of intravitreal bevacizumab and triamcinolone acetonide on persistent diffuse diabetic macular edema. Am. J. Ophthalmol. 2008, 145, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Moromizato, Y.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am. J. Pathol. 2000, 156, 1733–1739. [Google Scholar] [CrossRef]
- Das, A.; McGuire, P.G.; Monickaraj, F. Novel pharmacotherapies in diabetic retinopathy: Current status and what’s in the horizon? Indian J. Ophthalmol. 2016, 64, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Bucolo, C.; Leggio, G.M.; Drago, F.; Govoni, S.; Pascale, A. The PKCbeta/HuR/VEGF pathway in diabetic retinopathy. Biochem. Pharmacol. 2010, 80, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of phospholipases A2 in diabetic retinopathy: In vitro and in vivo studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Pascale, A.; Cupri, S.; Pignatello, R.; Osera, C.; Agata, V.D.; Amico, A.G.D.; Leggio, G.M.; Ruozi, B.; Govoni, S.; et al. Nanosystems based on siRNA silencing HuR expression counteract diabetic retinopathy in rat. Pharmacol. Res. 2016, 111, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Sone, H.; Okuda, Y.; Kawakami, Y.; Hanatani, M.; Suzuki, H.; Kozawa, T.; Honmura, S.; Yamashita, K. Vascular endothelial growth factor level in aqueous humor of diabetic patients with rubeotic glaucoma is markedly elevated. Diabetes Care 1996, 19, 1306–1307. [Google Scholar] [CrossRef] [PubMed]
- Dell’Omo, R.; Semeraro, F.; Bamonte, G.; Cifariello, F.; Romano, M.R.; Costagliola, C. Vitreous mediators in retinal hypoxic diseases. Mediat. Inflamm. 2013, 2013, 935301. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Bartoli, M.; Behzadian, M.A.; El-Remessy, A.E.B.; Al-Shabrawey, M.; Platt, D.H.; Caldwell, R.W. Vascular endothelial growth factor and diabetic retinopathy: Pathophysiological mechanisms and treatment perspectives. Diabetes Metab. Res. Rev. 2003, 19, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Ponnalagu, M.; Subramani, M.; Jayadev, C.; Shetty, R.; Das, D. Retinal pigment epithelium-secretome: A diabetic retinopathy perspective. Cytokine 2017, 95, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Noma, H.; Mimura, T.; Eguchi, S.; Hori, S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 2009, 116, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.R.; Kim, Y.H.; Kim, S.Y.; Nam, G.Y.; Cheon, H.J.; Lee, S.J. Plasma concentrations of vascular endothelial growth factor in retinopathy of prematurity afetr intravitreal Bevacizumab injection. Retina 2015, 35, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; McGuire, P.G.; Franco Nitta, C.; Monickaraj, F.; Oruganti, S.R.; Das, A. Chemokine mediated monocyte trafficking into the retina: Role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS ONE 2014, 9, e108508. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Grigoropoulos, V.; Emfietzoglou, I.; Theodossiadis, G.; Tentolouris, N.; Delicha, E.; Katsiari, C.; Alexiadou, K.; Hatziagelaki, E.; Theodossiadis, P.G. Infliximab for diabetic macular edema refractory to laser photocoagulation: A randomized, double-blind, placebo-controlled, crossover, 32-week study. Diabetes Care 2010, 33, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Mocan, M.C.; Kadayifcilar, S.; Eldem, B. Elevated intravitreal interleukin-6 levels in patients with proliferative diabetic retinopathy. Can. J. Ophthalmol. J. Can. Ophtalmol. 2006, 41, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Yamashita, H.; Noma, H.; Mimura, T.; Nakamura, S.; Sakata, K.; Hori, S. Aqueous humor levels of cytokines are related to vitreous levels and progression of diabetic retinopathy in diabetic patients. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Elner, S.G.; Elner, V.M.; Jaffe, G.J.; Stuart, A.; Kunkel, S.L.; Strieter, R.M. Cytokines in proliferative diabetic retinopathy and proliferative vitreoretinopathy. Curr. Eye Res. 1995, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.T.; Samples, J.R.; Hefeneider, S.H.; Howes, E.L. Ocular inflammatory effects of intravitreal interleukin 1. Arch. Ophthalmol. 1987, 105, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.A.; Mohr, S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 2007, 56, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-Q.; Wang, Y.-S. The role of Toll-like receptors in retinal ischemic diseases. Int. J. Ophthalmol. 2016, 9, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M. Diabetic retinopathy and dysregulated innate immunity. Vis. Res. 2017, 139, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Simó-Servat, O.; Hernández, C.; Simó, R. Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediat. Inflamm. 2012, 2012, 872978. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Ki-I, Y.; Hashiguchi, T.; Kawahara, K.; Biswas, K.K.; Nakamura, M.; Sonoda, Y.; Yamakiri, K.; Okubo, A.; Sakamoto, T.; et al. Intraocular expression and release of high-mobility group box 1 protein in retinal detachment. Lab. Investig. J. Tech. Methods Pathol. 2009, 89, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Keino, H.; Sato, Y.; Kudo, A.; Kawakami, H.; Okada, A.A. High mobility group box protein-1 in experimental autoimmune uveoretinitis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2283–2290. [Google Scholar] [CrossRef] [PubMed]
- Jakus, V.; Rietbrock, N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol. Res. 2004, 53, 131–142. [Google Scholar] [PubMed]
- Santos, A.R.C.; Dvoriantchikova, G.; Li, Y.; Mohammad, G.; Abu El-Asrar, A.M.; Wen, R.; Ivanov, D. Cellular mechanisms of high mobility group 1 (HMGB-1) protein action in the diabetic retinopathy. PLoS ONE 2014, 9, e87574. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Zhang, X.-D.; Li, Y.-Y.; Chen, X.-M.; Tang, D.-R.; Ran, R.-J. Involvement of HMGB1 mediated signalling pathway in diabetic retinopathy: Evidence from type 2 diabetic rats and ARPE-19 cells under diabetic condition. Br. J. Ophthalmol. 2013, 97, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, U.; Jialal, I. Hyperglycemia induces Toll-like receptor-2 and -4 expression and activity in human microvascular retinal endothelial cells: Implications for diabetic retinopathy. J. Diabetes Res. 2014, 2014, 790902. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-R.; Li, Z.-J.; Zeng, P.; Lan, Y.-Q. TLR7 deficiency contributes to attenuated diabetic retinopathy via inhibition of inflammatory response. Biochem. Biophys. Res. Commun. 2017, 493, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Platania, C.B.M.; Giurdanella, G.; Di Paola, L.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem. Pharmacol. 2017, 138, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Dinarello, C.A. Osmotic regulation of cytokine synthesis in vitro. Proc. Natl. Acad. Sci. USA 1995, 92, 12230–12234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, C.; Cao, S.; Ye, Z.; Deng, B.; Kijlstra, A.; Yang, P. High-salt enhances the inflammatory response by retina pigment epithelium cells following lipopolysaccharide stimulation. Mediat. Inflamm. 2015, 2015, 197521. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Quast, T.; Schröder, A.; Hucke, S.; Klotz, L.; Jantsch, J.; Gerzer, R.; Hemmersbach, R.; Kolanus, W. Salt-dependent chemotaxis of macrophages. PLoS ONE 2013, 8, e73439. [Google Scholar] [CrossRef]
- Schorn, C.; Frey, B.; Lauber, K.; Janko, C.; Strysio, M.; Keppeler, H.; Gaipl, U.S.; Voll, R.E.; Springer, E.; Munoz, L.E.; et al. Sodium overload and water influx activate the NALP3 inflammasome. J. Biol. Chem. 2011, 286, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Medzhitov, R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 2015, 6, 6931. [Google Scholar] [CrossRef] [PubMed]
- Binger, K.J.; Gebhardt, M.; Heinig, M.; Rintisch, C.; Schroeder, A.; Neuhofer, W.; Hilgers, K.; Manzel, A.; Schwartz, C.; Kleinewietfeld, M.; et al. High salt reduces the activation of IL4- and IL13-stimulated macrophages. J. Clin. Investig. 2015, 125, 4223–4238. [Google Scholar] [CrossRef] [PubMed]
- Yi, B.; Titze, J.; Rykova, M.; Feuerecker, M.; Vassilieva, G.; Nichiporuk, I.; Schelling, G.; Morukov, B.; Choukèr, A. Effects of dietary salt levels on monocytic cells and immune responses in healthy human subjects: A longitudinal study. Transl. Res. J. Lab. Clin. Med. 2015, 166, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Binger, K.J.; Linker, R.A.; Muller, D.N.; Kleinewietfeld, M. Sodium chloride, SGK1, and Th17 activation. Pflügers Archiv 2015, 467, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Jörg, S.; Grohme, D.A.; Erzler, M.; Binsfeld, M.; Haghikia, A.; Müller, D.N.; Linker, R.A.; Kleinewietfeld, M. Environmental factors in autoimmune diseases and their role in multiple sclerosis. Cell. Mol. Life Sci. 2016, 73, 4611–4622. [Google Scholar] [CrossRef] [PubMed]
- Hammer, A.; Schliep, A.; Jörg, S.; Haghikia, A.; Gold, R.; Kleinewietfeld, M.; Müller, D.N.; Linker, R.A. Impact of combined sodium chloride and saturated long-chain fatty acid challenge on the differentiation of T helper cells in neuroinflammation. J. Neuroinflamm. 2017, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Sigaux, J.; Semerano, L.; Favre, G.; Bessis, N.; Boissier, M.-C. Salt, inflammatory joint disease, and autoimmunity. Jt. Bone Spine 2017. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.L.; Kitz, A.; Wu, C.; Lowther, D.E.; Rodriguez, D.M.; Vudattu, N.; Deng, S.; Herold, K.C.; Kuchroo, V.K.; Kleinewietfeld, M.; et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Investig. 2015, 125, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-Z.; Le, Y.-Z. Significance of outer blood-retina barrier breakdown in diabetes and ischemia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Omri, S.; Behar-Cohen, F.; Rothschild, P.-R.; Gélizé, E.; Jonet, L.; Jeanny, J.C.; Omri, B.; Crisanti, P. PKCζ mediates breakdown of outer blood-retinal barriers in diabetic retinopathy. PLoS ONE 2013, 8, e81600. [Google Scholar] [CrossRef] [PubMed]
- Prager, P.; Hollborn, M.; Steffen, A.; Wiedemann, P.; Kohen, L.; Bringmann, A. P2Y1 receptor signaling contributes to high salt-induced priming of the NLRP3 inflammasome in retinal pigment epithelial cells. PLoS ONE 2016, 11, e0165653. [Google Scholar] [CrossRef] [PubMed]
- Atchison, E.; Barkmeier, A. The role of systemic risk factors in diabetic retinopathy. Curr. Ophthalmol. Rep. 2016, 4, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Ferraris, J.D.; Dmitrieva, N.I. Cellular response to hyperosmotic stresses. Physiol. Rev. 2007, 87, 1441–1474. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef] [PubMed]
- Neuhofer, W. Role of NFAT5 in inflammatory disorders associated with osmotic stress. Curr. Genom. 2010, 11, 584–590. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adorante, J.S.; Miller, S.S. Potassium-dependent volume regulation in retinal pigment epithelium is mediated by Na, K, Cl cotransport. J. Gen. Physiol. 1990, 96, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Civan, M.M.; Marano, C.W.; Matschinsky, F.W.; Peterson-Yantorno, K. Prolonged incubation with elevated glucose inhibits the regulatory response to shrinkage of cultured human retinal pigment epithelial cells. J. Membr. Biol. 1994, 139, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.R.; Carper, D.; Yokoyama, T.; Reddy, V.N. The effect of hypertonicity on aldose reductase, alpha B-crystallin, and organic osmolytes in the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2352–2359. [Google Scholar]
- Sato, S.; Lin, L.R.; Reddy, V.N.; Kador, P.F. Aldose reductase in human retinal pigment epithelial cells. Exp. Eye Res. 1993, 57, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Henry, D.N.; Frank, R.N.; Hootman, S.R.; Rood, S.E.; Heilig, C.W.; Busik, J.V. Glucose-specific regulation of aldose reductase in human retinal pigment epithelial cells in vitro. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1554–1560. [Google Scholar]
- Libert, S.; Willermain, F.; Weber, C.; Bryla, A.; Salik, D.; Gregoire, F.; Bolaky, N.; Caspers, L.; Perret, J.; Delporte, C. Involvement of TonEBP/NFAT5 in osmoadaptative response of human retinal pigmented epithelial cells to hyperosmolar stress. Mol. Vis. 2016, 22, 100–115. [Google Scholar] [PubMed]
- Winges, A.; Garcia, T.B.; Prager, P.; Wiedemann, P.; Kohen, L.; Bringmann, A.; Hollborn, M. Osmotic expression of aldose reductase in retinal pigment epithelial cells: Involvement of NFAT5. Graefes Arch. Clin. Exp. Ophthalmol. 2016, 254, 2387–2400. [Google Scholar] [CrossRef] [PubMed]
- Kador, P.F.; Wyman, M.; Oates, P.J. Aldose reductase, ocular diabetic complications and the development of topical Kinostat®. Prog. Retin. Eye Res. 2016, 54, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Grewal, A.S.; Bhardwaj, S.; Pandita, D.; Lather, V.; Sekhon, B.S. Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Rev. Med. Chem. 2016, 16, 120–162. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rivera, F.; Concheiro, A.; Alvarez-Lorenzo, C. Epalrestat-loaded silicone hydrogels as contact lenses to address diabetic-eye complications. Eur. J. Pharm. Biopharm. 2018, 122, 126–136. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbeny, A.; Naggar, H.; Miyauchi, S.; Ola, M.S.; Maddox, D.M.; Martin, P.M.; Ganapathy, V.; Smith, S.B. Osmoregulation of taurine transporter function and expression in retinal pigment epithelial, ganglion, and müller cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 694–701. [Google Scholar] [CrossRef][Green Version]
- Park, J.; Kim, H.; Park, S.Y.; Lim, S.W.; Kim, Y.S.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; et al. Tonicity-responsive enhancer binding protein regulates the expression of aldose reductase and protein kinase C δ in a mouse model of diabetic retinopathy. Exp. Eye Res. 2014, 122, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Reichmuth, K.; Prager, P.; Wiedemann, P.; Bringmann, A.; Kohen, L. Osmotic induction of placental growth factor in retinal pigment epithelial cells in vitro: Contribution of NFAT5 activity. Mol. Biol. Rep. 2016, 43, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Orgül, S.; Reuter, U.; Kain, H.L. Osmotic stress in an in vitro model of the outer blood-retinal barrier. Ger. J. Ophthalmol. 1993, 2, 436–443. [Google Scholar] [PubMed]
- Yamada, M.; Suzuki, E.; Kikuchi, G.; Hamazaki, J.; Hamazaki, S.; Matsuo, H. The hyperosmolarity-induced response of the ocular standing potential in mature rabbits. Doc. Ophthalmol. Adv. Ophthalmol. 1987, 66, 347–358. [Google Scholar] [CrossRef]
- Mukoh, S.; Kawasaki, K.; Yonemura, D.; Tanabe, J. Hyperosmolarity-induced hyperpolarization of the membrane potential of the retinal pigment epithelium. Doc. Ophthalmol. Adv. Ophthalmol. 1985, 60, 369–374. [Google Scholar] [CrossRef]
- Shirao, Y.; Steinberg, R.H. Mechanisms of effects of small hyperosmotic gradients on the chick RPE. Investig. Ophthalmol. Vis. Sci. 1987, 28, 2015–2025. [Google Scholar]
- Arsenijevic, T.; Vujovic, A.; Libert, F.; Op de Beeck, A.; Hébrant, A.; Janssens, S.; Grégoire, F.; Lefort, A.; Bolaky, N.; Perret, J.; et al. Hyperosmotic stress induces cell cycle arrest in retinal pigmented epithelial cells. Cell Death Dis. 2013, 4, e662. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Fujiseki, Y.; Omori, K.; Suzukawa, J.; Inagaki, C. Regulation of the expression of lysyl oxidase mRNA in cultured rabbit retinal pigment epithelium cells. Matrix Biol. J. Int. Soc. Matrix Biol. 2002, 21, 337–348. [Google Scholar] [CrossRef]
- Veltmann, M.; Hollborn, M.; Reichenbach, A.; Wiedemann, P.; Kohen, L.; Bringmann, A. Osmotic induction of angiogenic growth factor expression in human retinal pigment epithelial cells. PLoS ONE 2016, 11, e0147312. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
{kind=link}
| Layer’s No. | Layer’s Name | Layer’s Cell Types |
|---|---|---|
| 1 | Inner limiting membrane (ILM) | Müller cells (endfeet) |
| Astrocytes | ||
| 2 | Nerve fiber layer (NFL) | Ganglion cells (axons) |
| Retinal blood vessels cells | ||
| Glial cells | ||
| 3 | Ganglion cell layer (GCL) | Ganglion cells (nucleus) |
| Retinal blood vessels cells | ||
| Glial cells | ||
| Amacrine cells | ||
| 4 | Inner plexiform layer (IPL) | Bipolar cells |
| Ganglion cells | ||
| Amacrine cells | ||
| 5 | Inner nuclear layer (INL) | Bipolar cells (nucleus) |
| Horizontal cells (nucleus) | ||
| Amacrine cells (nucleus) | ||
| Müller cells (nucleus) | ||
| 6 | Outer plexiform layer (OPL) | Photoreceptor cells |
| Bipolar cells | ||
| Horizontal cells | ||
| 7 | Outer nuclear layer (ONL) | Photoreceptor cells (nucleus) |
| 8 | Outer limiting membrane (OLM) | Photoreceptor cells |
| Müller cells | ||
| 9 | Photoreceptor layer (PL) | Photoreceptor cells (rods and cones) |
| 10 | Retinal pigmented epithelium (RPE) | Retinal pigmented epithelial cells |
| AQP | Rat RPE | Human RPE |
|---|---|---|
| AQP0 | [32,33] | - |
| AQP1 | [32,33,34,35] | [36,37,38,39] controversy |
| AQP2 | [34] | |
| AQP3 | [33] | [40] |
| AQP4 | [34,35,41] | - |
| AQP5 | [32,33] | [42] |
| AQP6 | [33,34] | - |
| AQP7 | [33] | [43] |
| AQP8 | [33] | [42] |
| AQP9 | [32] | - |
| AQP10 | - | - |
| AQP11 | [32,33] | - |
| AQP12 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willermain, F.; Scifo, L.; Weber, C.; Caspers, L.; Perret, J.; Delporte, C. Potential Interplay between Hyperosmolarity and Inflammation on Retinal Pigmented Epithelium in Pathogenesis of Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 1056. https://doi.org/10.3390/ijms19041056
Willermain F, Scifo L, Weber C, Caspers L, Perret J, Delporte C. Potential Interplay between Hyperosmolarity and Inflammation on Retinal Pigmented Epithelium in Pathogenesis of Diabetic Retinopathy. International Journal of Molecular Sciences. 2018; 19(4):1056. https://doi.org/10.3390/ijms19041056
Chicago/Turabian StyleWillermain, François, Lisa Scifo, Célia Weber, Laure Caspers, Jason Perret, and Christine Delporte. 2018. "Potential Interplay between Hyperosmolarity and Inflammation on Retinal Pigmented Epithelium in Pathogenesis of Diabetic Retinopathy" International Journal of Molecular Sciences 19, no. 4: 1056. https://doi.org/10.3390/ijms19041056
APA StyleWillermain, F., Scifo, L., Weber, C., Caspers, L., Perret, J., & Delporte, C. (2018). Potential Interplay between Hyperosmolarity and Inflammation on Retinal Pigmented Epithelium in Pathogenesis of Diabetic Retinopathy. International Journal of Molecular Sciences, 19(4), 1056. https://doi.org/10.3390/ijms19041056