The Role of Single-Molecule Force Spectroscopy in Unraveling Typical and Autoimmune Heparin-induced Thrombocytopenia


Abstract
1. Introduction
2. Insights into Binding Mechanisms of Typical HIT
2.1. Heparins and PF4 Associated with HIT
2.2. Boundary between Antigenic and Nonantigenic Heparins
2.3. Long Heparin-Induced Additional Bond among PF4 Molecules
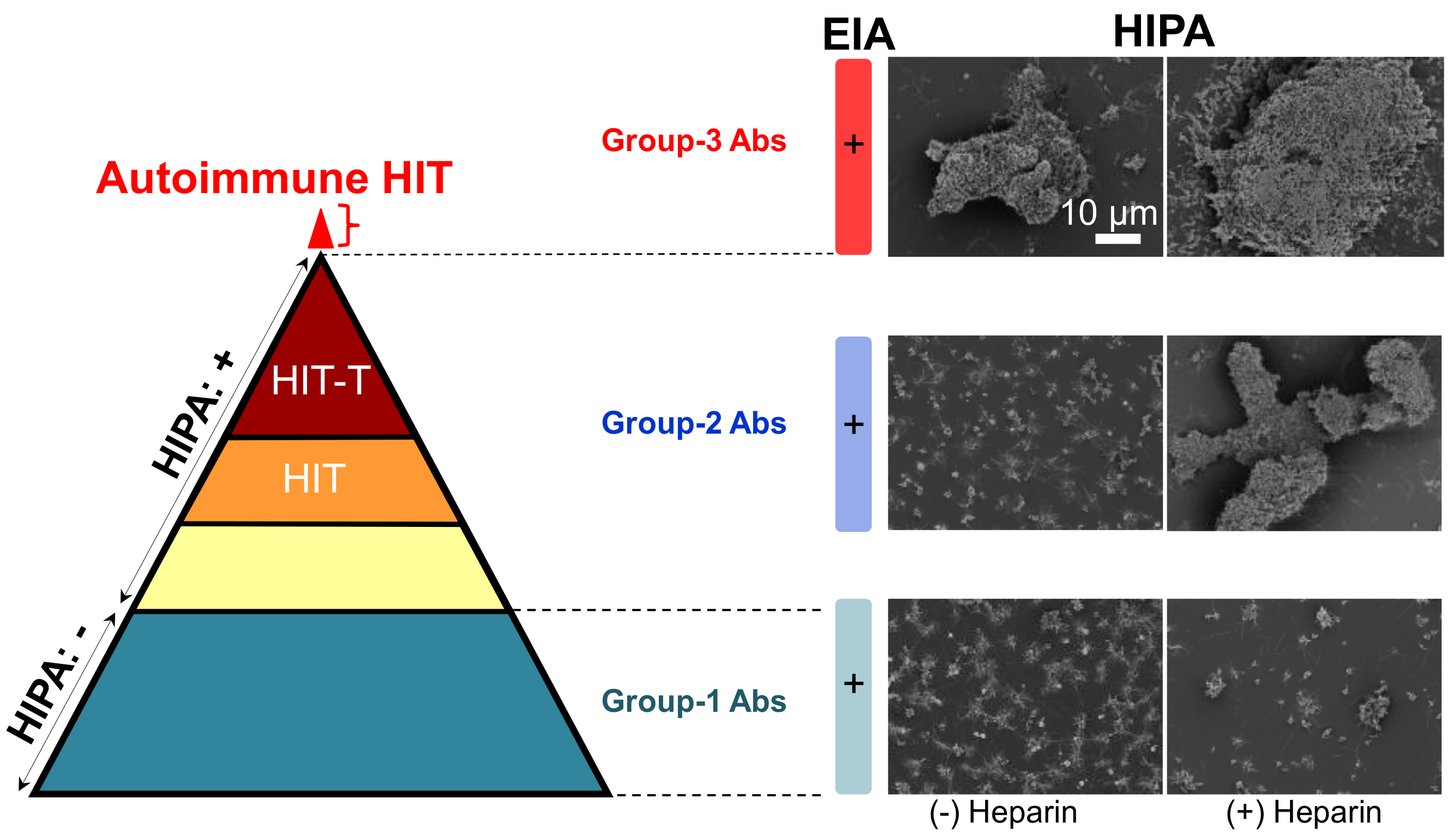
3. Insights into Binding Mechanism of aPF4/H Abs-Induced Autoimmune HIT
3.1. Characteristics of aPF4/H Abs
3.2. Bond Dynamics of aPF4/H Abs
3.3. Autoimmune HIT (Group-3) Antibodies Cluster PF4 and Allow Binding of Other aPF4/H Abs
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| HIT | Heparin-induced thrombocytopenia |
| PF4 | Platelet factor 4 |
| H | Heparin |
| PF4/H | Platelet factor 4/Heparin |
| aPF4/H Abs | anti-PF4/Heparin antibodies |
| PF4/P | Platelet factor 4/Polyanion |
| HO05 | Fondaparinux |
| HO06 | 6-mer heparin |
| HO08 | 8-mer heparin |
| HO12 | 12-mer heparin |
| HO16 | 16-mer heparin |
| UFH | Unfractionated heparin |
| KKO | Mouse monoclonal antibody that recognizes human platelet factor 4 |
| AFM | Atomic force microscopy |
| SMFS | Single-molecule force spectroscopy |
| ELISA | Enzyme-linked immunosorbent assay |
| PEG | Polyethylene glycol |
| HIPA | Platelet activation assay |
| DLS | Dynamic light scattering |
| ITC | Isothermal titration calorimetry |
| CD | Circular dichroism |
| OD | Optical density |
References
- Collins, J.; Greinacher, A.; MacCallum, P. Autoimmune heparin-induced thrombocytopenia: A case report. Br. J. Haematol. 2016, 173, 169. [Google Scholar]
- Nguyen, T.H. Single-molecule force spectroscopy applied to heparin-induced thrombocytopenia. J. Mol. Recognit 2016. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; van Boeckel, C.A. A synthetic antithrombin iii binding pentasaccharide is now a drug! What comes next? Angew. Chem. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. The growing complexity of platelet aggregation. Blood 2007, 109, 5087–5095. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Krauel, K.; Gottschalk, K.E.; Renne, T.; Helm, C.A.; Greinacher, A.; Block, S. Characterisation of the conformational changes in platelet factor 4 induced by polyanions: Towards in vitro prediction of antigenicity. Thromb. Haemost. 2014, 112, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A. Heparin-induced thrombocytopenia. N. Engl. J. Med. 2015, 373, 1883–1884. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Greinacher, A. Heparin-Induced Thrombocytopenia, 4th ed.; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Blank, M.; Shoenfeld, Y.; Tavor, S.; Praprotnik, S.; Boffa, M.C.; Weksler, B.; Walenga, M.J.; Amiral, J.; Eldor, A. Anti-platelet factor 4/heparin antibodies from patients with heparin-induced thrombocytopenia provoke direct activation of microvascular endothelial cells. Int. Immunol. 2002, 14, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kelton, J.G.; Sheridan, D.; Santos, A.; Smith, J.; Steeves, K.; Smith, C.; Brown, C.; Murphy, W.G. Heparin-induced thrombocytopenia-laboratory studies. Blood 1988, 72, 925–930. [Google Scholar] [PubMed]
- Rollin, J.; Pouplard, C.; Gruel, Y. Risk factors for heparin-induced thrombocytopenia: Focus on fc gamma receptors. Thromb. Haemost. 2016, 116, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia. J. Thromb. Haemost. 2017, 15, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Whiteheart, S.W. Platelet granules: Surprise packages. Blood 2011, 118, 1190–1191. [Google Scholar] [CrossRef] [PubMed]
- Sixma, J.J.; Wester, J. The hemostatic plug. Semin. Hematol. 1977, 14, 265–299. [Google Scholar] [PubMed]
- Block, S.; Greinacher, A.; Helm, C.A.; Delcea, M. Characterization of bonds formed between platelet factor 4 and negatively charged drugs using single molecule force spectroscopy. Soft Matter 2014, 10, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Kreimann, M.; Brandt, S.; Krauel, K.; Block, S.; Helm, C.A.; Weitschies, W.; Greinacher, A.; Delcea, M. Binding of anti-platelet factor 4/heparin antibodies depends on the thermodynamics of conformational changes in platelet factor 4. Blood 2014, 124, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Heddle, N.M. Laboratory diagnosis of immune heparin-induced thrombocytopenia. Curr. Hematol. Rep. 2003, 2, 148–157. [Google Scholar] [PubMed]
- Verma, A.K.; Levine, M.; Shalansky, S.J.; Carter, C.J.; Kelton, J.G. Frequency of heparin-induced thrombocytopenia in critical care patients. Pharmacotherapy 2003, 23, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.A.; Cook, D.J.; Albert, M.; Williamson, D.; Meade, M.; Granton, J.; Skrobik, Y.; Langevin, S.; Mehta, S.; Hebert, P.; et al. The 4ts scoring system for heparin-induced thrombocytopenia in medical-surgical intensive care unit patients. J. Crit. Care 2010, 25, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Jay, R.M.; Makris, M.; Kelton, J.G. Platelet-activating anti-platelet factor 4/polyanion antibodies without preceding heparin therapy: A transient autoimmune disorder resembling heparin-induced thrombocytopenia (“spontaneous hit”). Blood 2006, 108, 311a–312a. [Google Scholar]
- Shoenfeld, Y. Heparin-induced thrombocytopenia as an autoimmune disease-idiotypic evidence for the role of anti-heparin/pf4 autoantibodies. Isr. J. Med. Sci. 1997, 33, 243–245. [Google Scholar] [PubMed]
- Nguyen, T.H.; Greinacher, A.; Delcea, M. Quantitative description of thermodynamic and kinetic properties of the platelet factor 4/heparin bonds. Nanoscale 2015, 7, 10130–10139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-platelet factor 4/polyanion antibodies mediate a new mechanism of autoimmunity. Nat. Commun. 2017, 8, 14945. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.A.G.A. Platelet factor 4/heparin complexes present their epitopes differently on a solid phase system than on the platelet surface. Blood 2017. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.; Lindsay, S.M.; Leuba, S.H. Single molecule force spectroscopy in biology using the atomic force microscope. Prog. Biophys. Mol. Biol. 2000, 74, 37–61. [Google Scholar] [CrossRef]
- Neuman, K.C.; Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods 2008, 5, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Hinterdorfer, P.; Dufrene, Y.F. Detection and localization of single molecular recognition events using atomic force microscopy. Nat. Methods 2006, 3, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, A.R.; Cannistraro, S. The application of atomic force spectroscopy to the study of biological complexes undergoing a biorecognition process. Chem. Soc. Rev. 2010, 39, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Casuso, I.; Rico, F.; Scheuring, S. Biological afm: Where we come from—where we are—where we may go. J. Mol. Recognit. 2011, 24, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.J.; Helenius, J.; Alsteens, D.; Dufrene, Y.F. Force probing surfaces of living cells to molecular resolution. Nat. Chem. Biol. 2009, 5, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Chemla, Y.R.; Smith, S.B.; Bustamante, C. Recent advances in optical tweezers. Annu. Rev. Biochem. 2008, 77, 205–228. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, R.I.; Yarovoi, S.V.; Rauova, L.; Barsegov, V.; Sachais, B.S.; Rux, A.H.; Hinds, J.L.; Arepally, G.M.; Cines, D.B.; Weisel, J.W. Distinct specificity and single-molecule kinetics characterize the interaction of pathogenic and non-pathogenic antibodies against platelet factor 4-heparin complexes with platelet factor 4. J. Biol. Chem. 2013, 288, 33060–33070. [Google Scholar] [CrossRef] [PubMed]
- Dufrene, Y.F.; Hinterdorfer, P. Recent progress in afm molecular recognition studies. Pfluger Arch. Eur. J. Phys. 2008, 456, 237–245. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nguyen, T.H.; Kim, Y.U.; Kim, K.J.; Choi, S.S. Investigation of structural transition of dsdna on various substrates studied by atomic force microscopy. J. Nanosci. Nanotechnol. 2009, 9, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Lee, S.M.; Na, K.; Yang, S.; Kim, J.; Yoon, E.S. An improved measurement of dsdna elasticity using afm. Nanotechnology 2010, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Wojcikiewicz, E.; Moy, V.T. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys. J. 2002, 83, 2270–2279. [Google Scholar] [CrossRef]
- Carvalho, F.A.; Connell, S.; Miltenberger-Miltenyi, G.; Pereira, S.V.; Tavares, A.; Ariens, R.A.S.; Santos, N.C. Atomic force microscopy-based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano 2010, 4, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, J.; Legate, K.R.; Schubert, R.; Bharadwaj, M.; Werner, C.; Mullner, D.J.; Benoit, M. A practical guide to quantify cell adhesion using single-cell force spectroscopy. Methods 2013, 60, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Alsteens, D.; Van Dijck, P.; Lipke, P.N.; Dufrene, Y.F. Quantifying the forces driving cell-cell adhesion in a fungal pathogen. Langmuir 2013, 29, 13473–13480. [Google Scholar] [CrossRef] [PubMed]
- Beaussart, A.; El-Kirat-Chatel, S.; Sullan, R.M.A.; Alsteens, D.; Herman, P.; Derclaye, S.; Dufrene, Y.F. Quantifying the forces guiding microbial cell adhesion using single-cell force spectroscopy. Nat. Protoc. 2014, 9, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Gabriel, D.; Gerisch, G.; Gaub, H.E. Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat. Cell. Biol. 2000, 2, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Nguyen, T.H.; Na, K.; Cho, I.J.; Woo, D.H.; Oh, J.E.; Lee, C.J.; Yoon, E.S. Nanomechanical measurement of astrocyte stiffness correlated with cytoskeletal maturation. J. Biomed. Mater. Res. A 2015, 103, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.C.; Nguyen, T.H. The role of cd4 on mechanical properties of live cell membrane. J. Biomed. Mater. Res. A 2016, 104, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.A.; Santos, N.C. Atomic force microscopy-based force spectroscopy u biological and biomedical applications. Iubmb Life 2012, 64, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Ros, R.; Eckel, R.; Bartels, F.; Sischka, A.; Baumgarth, B.; Wilking, S.D.; Puhler, A.; Sewald, N.; Becker, A.; Anselmetti, D. Single molecule force spectroscopy on ligand-DNA complexes: From molecular binding mechanisms to biosensor applications. J. Biotechnol. 2004, 112, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Steinbock, L.J.; Butt, H.J.; Helm, M.; Berger, R. Measuring single small molecule binding via rupture forces of a split aptamer. J. Am. Chem. Soc. 2011, 133, 2025–2027. [Google Scholar] [CrossRef] [PubMed]
- Dupres, V.; Verbelen, C.; Dufrene, Y.F. Probing molecular recognition sites on biosurfaces using afm. Biomaterials 2007, 28, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Kelton, J.G. Heparin-induced thrombocytopenia. Annu. Rev. Med. 1989, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.K.; Yates, E.A.; Fernig, D.G.; Turnbull, J.E. Interactions of heparin/heparan sulfate with proteins: Appraisal of structural factors and experimental approaches. Glycobiology 2004, 14, 17R–30R. [Google Scholar] [CrossRef] [PubMed]
- Valle-Delgado, J.J.; Urban, P.; Fernandez-Busquets, X. Demonstration of specific binding of heparin to plasmodium falciparum-infected vs. Non-infected red blood cells by single-molecule force spectroscopy. Nanoscale 2013, 5, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Laremore, T.N.; Zhang, F.; Dordick, J.S.; Liu, J.; Linhardt, R.J. Recent progress and applications in glycosaminoglycan and heparin research. Curr. Opin. Chem. Biol. 2009, 13, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Marszalek, P.E.; Oberhauser, A.F.; Li, H.; Fernandez, J.M. The force-driven conformations of heparin studied with single molecule force microscopy. Biophys. J. 2003, 85, 2696–2704. [Google Scholar] [CrossRef]
- Lee, G.; Nowak, W.; Jaroniec, J.; Zhang, Q.M.; Marszalek, P.E. Molecular dynamics simulations of forced conformational transitions in 1,6-linked polysaccharides. Biophys. J. 2004, 87, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.T., Jr.; Ursick, J.A.; Heim, K.L.; Hilleman, D.E.; Reich, J.W. Heparin-associated thrombocytopenia: A prospective comparison of bovine lung heparin, manufactured by a new process, and porcine intestinal heparin. Drug Intell. Clin. Pharm. 1986, 20, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.L.; Palmer, G.J., 3rd; Moroose, R.; Drexler, A. Comparison of bovine and porcine heparin in heparin antibody formation after cardiac surgery. Ann. Thorac. Surg. 2003, 75, 17–22. [Google Scholar] [CrossRef]
- Green, D.; Martin, G.J.; Shoichet, S.H.; Debacker, N.; Bomalaski, J.S.; Lind, R.N. Thrombocytopenia in a prospective, randomized, double-blind trial of bovine and porcine heparin. Am. J. Med. Sci. 1984, 288, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lu, X.A.; Kulkarni, S.S.; Wen, Y.S.; Hung, S.C. Synthesis of heparin oligosaccharides. J. Am. Chem. Soc. 2004, 126, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Codee, J.D.C.; Stubba, B.; Schiattarella, M.; Overkleeft, H.S.; van Boeckel, C.A.A.; van Boom, J.H.; van der Marel, G.A. A modular strategy toward the synthesis of heparin-like oligosaccharides using monomeric building blocks in a sequential glycosylation strategy. J. Am. Chem. Soc. 2005, 127, 3767–3773. [Google Scholar] [CrossRef] [PubMed]
- De Paz, J.L.; Noti, C.; Seeberger, P.H. Microarrays of synthetic heparin oligosaccharides. J. Am. Chem. Soc. 2006, 128, 2766–2767. [Google Scholar] [CrossRef] [PubMed]
- Polat, T.; Wong, C.H. Anomeric reactivity-based one-pot synthesis of heparin-like oligosaccharides. J. Am. Chem. Soc. 2007, 129, 12795–12800. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.M.; Cai, C.; Chandarajoti, K.; Hsieh, P.H.; Li, L.Y.; Pham, T.Q.; Sparkenbaugh, E.M.; Sheng, J.Z.; Key, N.S.; Pawlinski, R.; et al. Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat. Chem. Biol. 2014, 10, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultralarge complexes of pf4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Levine, M.N.; Roberts, R.S.; Gent, M.; Horsewood, P.; Kelton, J.G. Heparin-induced thrombocytopenia is more common with unfractionated heparin than with low-molecular-weight heparin. Thromb. Haemost. 1993, 69, 911. [Google Scholar]
- Linhardt, R.J.; Liu, J. Synthetic heparin. Curr. Opin. Pharmacol. 2012, 12, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Maurer, B.T.; Aster, R.H. Heparin-induced thrombocytopenia associated with fondaparinux. N. Engl. J. Med. 2007, 356, 2653–2654. [Google Scholar] [PubMed]
- Martel, N.; Lee, J.; Wells, P.S. Risk for heparin-induced thrombocytopenia. With unfractionated and low-molecular-weight heparin thromboprophylaxis: A meta-analysis. Blood 2005, 106, 2710–2715. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.; Ritchie, K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997, 72, 1541–1555. [Google Scholar] [CrossRef]
- Saboury, A.A. A review on the ligand binding studies by isothermal titration calorimetry. J. Iran. Chem. Soc. 2006, 3, 1–21. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Heparin-induced thrombocytopenia. Curr. Opin. Crit. Care 2015, 21, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Greinacher, A.; Gruel, Y.; Aster, R.H.; Chong, B.H.; Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Laboratory testing for heparin-induced thrombocytopenia: A conceptual framework and implications for diagnosis. J. Thromb. Haemost. 2011, 9, 2498–2500. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Basciano, P.A.; Knopman, J.; Bernstein, R.A. Spontaneous heparin-induced thrombocytopenia syndrome: 2 new cases and a proposal for defining this disorder. Blood 2014, 123, 3651–3654. [Google Scholar] [CrossRef] [PubMed]
- Amiral, J.; Pouplard, C.; Vissac, A.M.; Walenga, J.M.; Jeske, W.; Gruel, Y. Affinity purification of heparin-dependent antibodies to platelet factor 4 developed in heparin-induced thrombocytopenia: Biological characteristics and effects on platelet activation. Br. J. Haematol. 2000, 109, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Jones, C.G.; Bougie, D.W.; Curtis, B.R.; McFarland, J.G.; Wang, D.M.; Aster, R.H. Heparin-independent, pf4-dependent binding of hit antibodies to platelets: Implications for hit pathogenesis. Blood 2015, 125, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Rauova, L.; Arepally, G.; Reilly, M.P.; McKenzie, S.E.; Sachais, B.S.; Poncz, M. Heparin-induced thrombocytopenia: An autoimmune disorder regulated through dynamic autoantigen assembly/disassembly. J. Clin. Apher. 2007, 22, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Sachais, B.S.; Litvinov, R.I.; Yarovoi, S.V.; Rauova, L.; Hinds, J.L.; Rux, A.H.; Arepally, G.M.; Poncz, M.; Cuker, A.; Weisel, J.W.; et al. Dynamic antibody-binding properties in the pathogenesis of hit. Blood 2012, 120, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Reilly, M.P.; Taylor, S.M.; Hartman, N.K.; Arepally, G.M.; Sachais, B.S.; Cines, D.B.; Poncz, M.; McKenzie, S.E. Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through fc gamma riia. Blood 2001, 98, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Arepally, G.M.; Kamei, S.; Park, K.S.; Kamei, K.; Li, Z.Q.; Siegel, D.L.; Kisiel, W.; Cines, D.B.; Poncz, M. Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood 2000, 95, 1533–1540. [Google Scholar] [PubMed]
- Nguyen, T.H.; Greinacher, A. Effect of ph and ionic strength on the binding strength of anti-pf4/polyanion antibodies. Eur. Biophys. J. 2017, 46, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H. Not Only Heparin but Also Antibody Induces Thrombocytopenia; Abrol, P., Ed.; InTech: London, UK, 2018. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | F(pN) | koff (s−1) | Cluster PF4 |
|---|---|---|---|
| KKO | 43.6 ± 8.8 | 2.2 | weak |
| Group-1 | 44.0 ± 8.1 | 15.6 | no |
| Group-2 | 60.6 ± 15.4 | 2.0 | weak |
| Group-3 | 72.4 ± 26.2 | 0.12 | strong |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bui, V.-C.; Nguyen, T.-H. The Role of Single-Molecule Force Spectroscopy in Unraveling Typical and Autoimmune Heparin-induced Thrombocytopenia. Int. J. Mol. Sci. 2018, 19, 1054. https://doi.org/10.3390/ijms19041054
Bui V-C, Nguyen T-H. The Role of Single-Molecule Force Spectroscopy in Unraveling Typical and Autoimmune Heparin-induced Thrombocytopenia. International Journal of Molecular Sciences. 2018; 19(4):1054. https://doi.org/10.3390/ijms19041054
Chicago/Turabian StyleBui, Van-Chien, and Thi-Huong Nguyen. 2018. "The Role of Single-Molecule Force Spectroscopy in Unraveling Typical and Autoimmune Heparin-induced Thrombocytopenia" International Journal of Molecular Sciences 19, no. 4: 1054. https://doi.org/10.3390/ijms19041054
APA StyleBui, V.-C., & Nguyen, T.-H. (2018). The Role of Single-Molecule Force Spectroscopy in Unraveling Typical and Autoimmune Heparin-induced Thrombocytopenia. International Journal of Molecular Sciences, 19(4), 1054. https://doi.org/10.3390/ijms19041054