Whole-Genome Comparison Reveals Heterogeneous Divergence and Mutation Hotspots in Chloroplast Genome of Eucommia ulmoides Oliver

Abstract
1. Introduction
2. Results
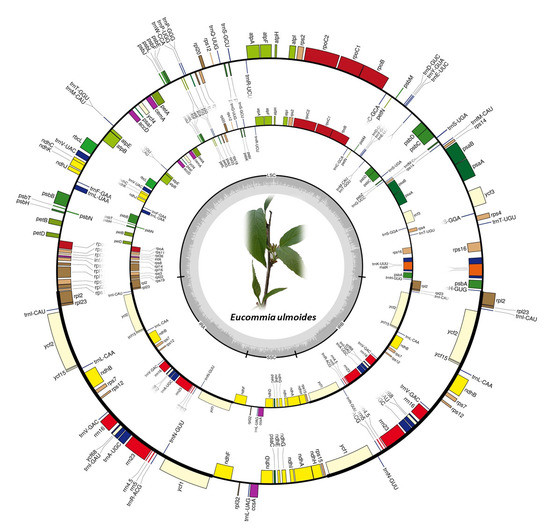
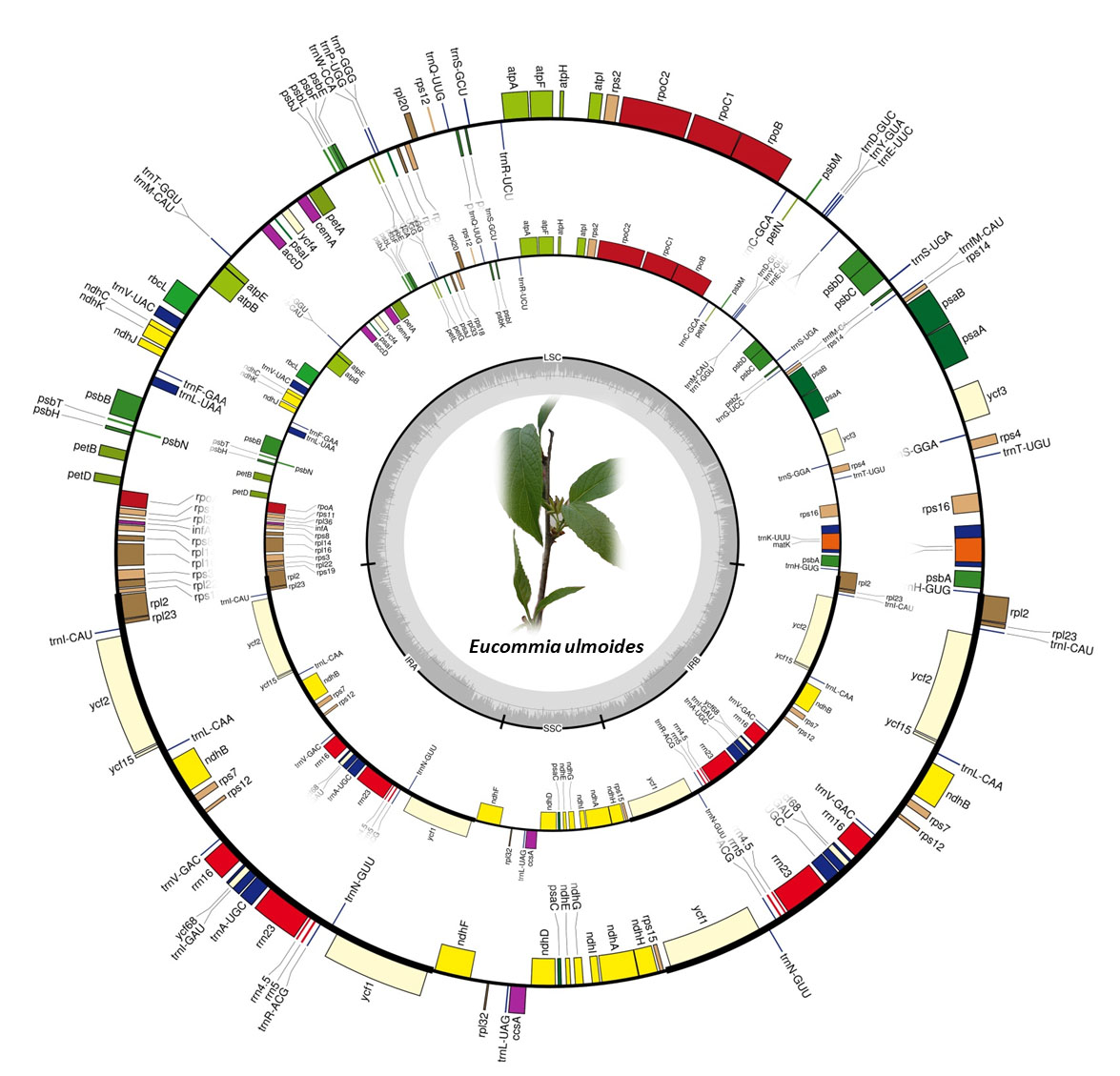
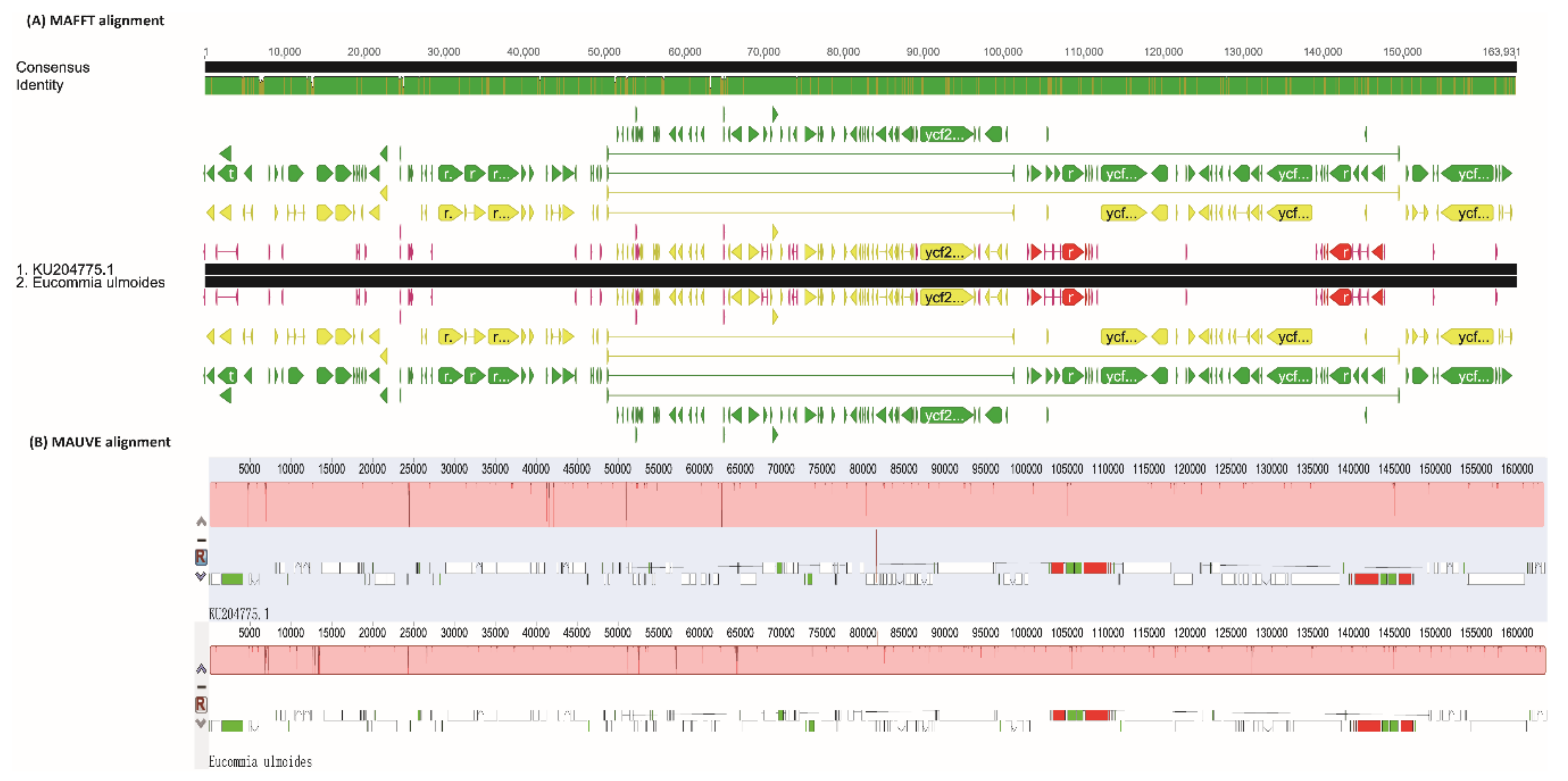
2.1. Chloroplast Genome Variation in E. ulmoides
2.2. Molecular Marker Development
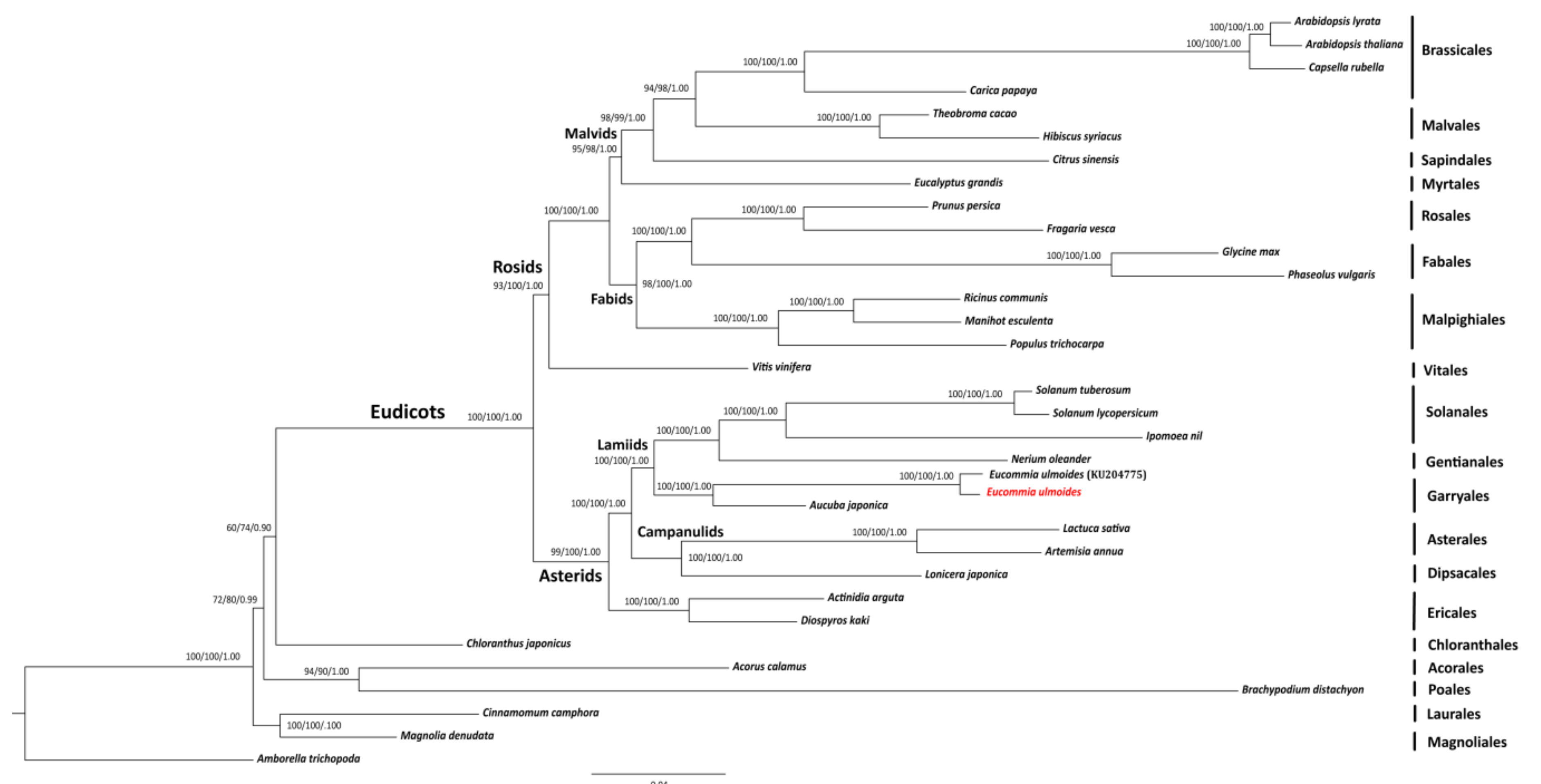
2.3. SNP Calling and Phylogenomic Inference
3. Discussion
3.1. Conserved Chloroplast Genome Structure in E. ulmoides
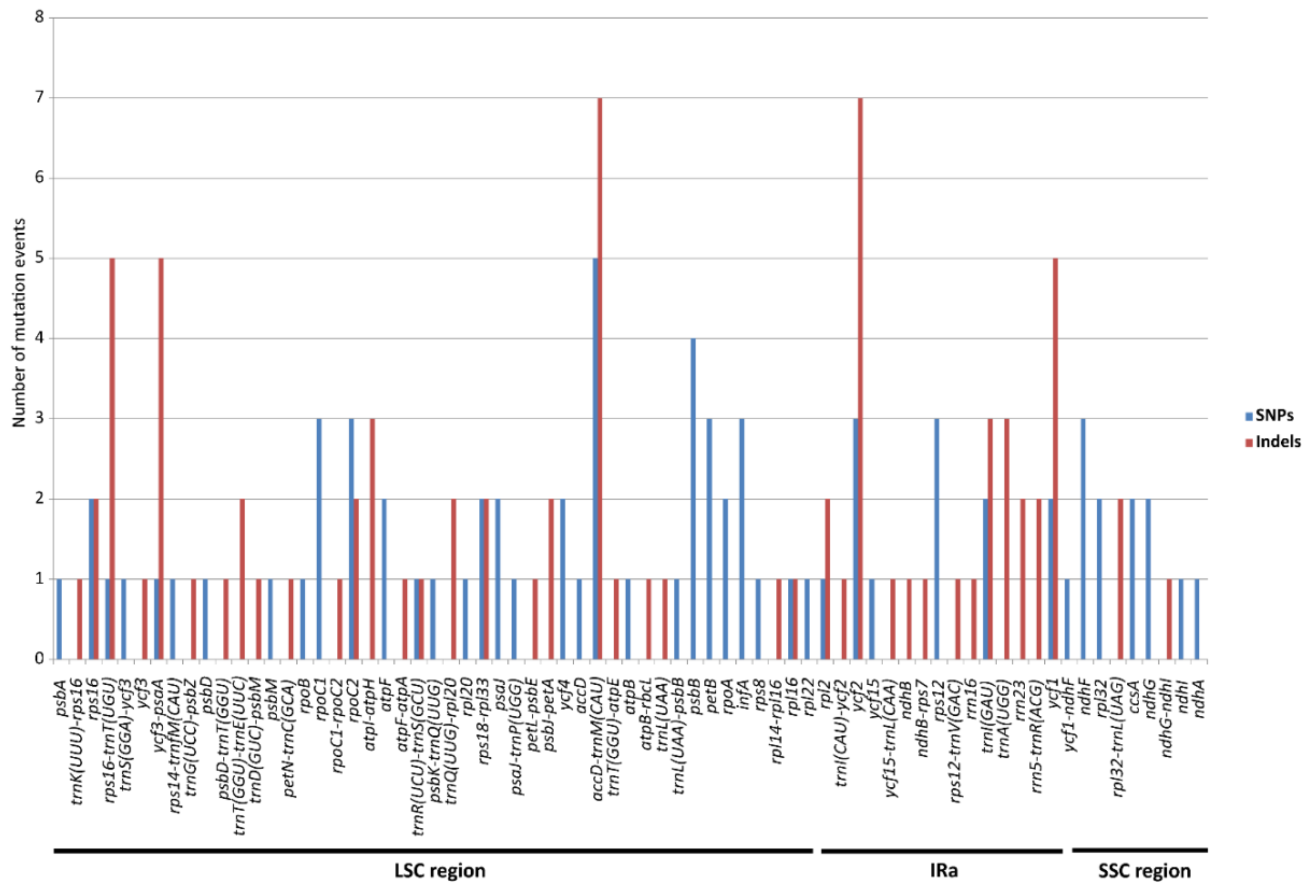
3.2. Heterogeneous Divergence in E. ulmoides Chloroplast Genome
3.3. Mutation Hotspots in E. ulmoides Chloroplast Genome
3.4. Phylogenomic Validation of E. ulmoides
4. Materials and Methods
4.1. Plant Materials and DNA Sequencing
4.2. Genome Assembly and Annotation
4.3. Genome-Wide Comparison and Divergent Hotspot Identification
4.4. SNPs Validation and Phylogenomic Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, H. Plant diversity and conservation in China: Planning a strategic bioresource for a sustainable future. Bot. J. Linn. Soc. 2011, 166, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ouyang, Z.; Pimm, S.L.; Raven, P.H.; Wang, X.; Miao, H.; Han, N. Protecting China’s biodiversity. Science 2003, 300, 1240–1241. [Google Scholar] [CrossRef] [PubMed]
- Lópezpujol, J.; Zhang, F.M.; Ge, S. Plant biodiversity in China: Richly varied, endangered, and in need of conservation. Biodivers. Conserv. 2006, 15, 3983–4026. [Google Scholar] [CrossRef]
- Gu, J. Conservation of plant diversity in China: Achievements, prospects and concerns. Biol. Conserv. 1998, 85, 321–327. [Google Scholar] [CrossRef]
- Chen, S.L.; Hua, Y.; Luo, H.M.; Wu, Q.; Li, C.F.; Steinmetz, A. Conservation and sustainable use of medicinal plants: Problems, progress, and prospects. Chin. Med. 2016, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, B.; Wang, M.; Fu, G. An approach to some problems on utilization of medicinal plant resource in China. China J. Chin. Mater. Med. 1999, 24, 70–73. [Google Scholar]
- Zhang, Z.Y.; Zhang, H.D.; Turland, N.J. Eucommiaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press and Missouri Botanical Garden: Beijing, China, 2003; p. 43. [Google Scholar]
- Kawasaki, T.; Uezono, K.; Nakazawa, Y. Antihypertensive mechanism of food for specified health use: “Eucommia leaf glycoside” and its clinical application. J. Health Sci. 2000, 22, 29–36. [Google Scholar]
- Liu, H.; Hongyan, D.U.; Tana, W. Advances in research on biotechnology breeding of Eucommia ulmoides. Hunan For. Sci. Technol. 2016, 43, 132–136. [Google Scholar]
- Suzuki, N.; Uefuji, H.; Nishikawa, T.; Mukai, Y.; Yamashita, A.; Hattori, M.; Ogasawara, N.; Bamba, T.; Fukusaki, E.; Kobayashi, A. Construction and analysis of EST libraries of the trans-polyisoprene producing plant, Eucommia ulmoides Oliver. Planta 2012, 236, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Du, H.Y.; Hu, W.Z.; Yu, R. The Report on Development of China’s Eucommia Rubber Resources and Industry (2014-2015); Social Sciences Academic Press: Beijing, China, 2015. [Google Scholar]
- Manchester, S.R.; Chen, Z.D.; An-Ming, L.U.; Uemura, K. Eastern Asian endemic seed plant genera and their paleogeographic history throughout the Northern Hemisphere. J. Syst. Evol. 2009, 47, 1–42. [Google Scholar] [CrossRef]
- Mabberley, D.J. The Plant Book; Cambridge University Press: Cambridge, UK, 1989. [Google Scholar]
- Fu, L.G.; Jin, J.M. Red List of Endangered Plants in China; Science Press: Beijing, China, 1992. [Google Scholar]
- Du, H.Y. China Eucommia Pictorial; China Forestry Publishing House: Beijing, China, 2014. [Google Scholar]
- Wang, L.; Du, H.; Wuyun, T.N. Genome-wide identification of microRNAs and their targets in the leaves and fruits of Eucommia ulmoides using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wuyun, T.N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Zhang, J.; Xing, C.; Tian, H.; Yao, X. Microsatellite genetic variation in the Chinese endemic Eucommia ulmoides (Eucommiaceae): Implications for conservation. Bot. J. Linn. Soc. 2013, 173, 775–785. [Google Scholar] [CrossRef][Green Version]
- Zhang, W.R.; Li, Y.; Zhao, J.; Wu, C.H.; Ye, S.; Yuan, W.J. Isolation and characterization of microsatellite markers for Eucommia ulmoides (Eucommiaceae), an endangered tree, using next-generation sequencing. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, D.; Li, Z.; Wei, J.; Jin, C.; Liu, M. A molecular genetic linkage map of Eucommia ulmoides and quantitative trait loci (QTL) analysis for growth traits. Int. J. Mol. Sci. 2014, 15, 2053–2074. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Y.; Li, L.; Wei, Y.; Li, Z. The first genetic linkage map of Eucommia ulmoides. J. Genet. 2014, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Q.; Huang, L.Q.; Shao, A.J.; Cui, G.H.; Chen, M.; Tong, C.H. Genetic diversity of Eucommia ulmoides by RAPD analysis. China J. Chin. Mater. Med. 2006, 31, 1583–1586. [Google Scholar]
- Yao, X.; Deng, J.; Huang, H. Genetic diversity in Eucommia ulmoides (Eucommiaceae), an endangered traditional Chinese medicinal plant. Conserv. Genet. 2012, 13, 1499–1507. [Google Scholar] [CrossRef]
- Yu, J.; Wang, Y.; Peng, L.; Ru, M.; Liang, Z.S. Genetic diversity and population structure of Eucommia ulmoides Oliver, an endangered medicinal plant in China. Genet. Mol. Res. 2015, 14, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Vignal, A.; Milan, D.; Sancristobal, M.; Eggen, A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 2002, 34, 275–305. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, J.; Mazza, R.; Caruso, P.; Reforgiato, R.G.; Marocco, A.; Licciardello, C. Use of an expressed sequence tag-based method for single nucleotide polymorphism identification and discrimination of Citrus species and cultivars. Mol. Breed. 2013, 31, 705–718. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Kess, T.; Gross, J.; Harper, F.; Boulding, E.G. Low-cost ddRAD method of SNP discovery and genotyping applied to the periwinkle Littorina saxatilis. J. Molluscan Stud. 2016, 82, eyv042. [Google Scholar]
- Potter, K.M.; Hipkins, V.D.; Mahalovich, M.F.; Means, R.E. Nuclear genetic variation across the range of ponderosa pine (Pinus ponderosa): Phylogeographic, taxonomic and conservation implications. Tree Genet. Genomes 2015, 11, 38. [Google Scholar] [CrossRef]
- Worth, J.R.P.; Yokogawa, M.; Pérez-Figueroa, A.; Tsumura, Y.; Tomaru, N.; Janes, J.K.; Isagi, Y. Conflict in outcomes for conservation based on population genetic diversity and genetic divergence approaches: A case study in the Japanese relictual conifer Sciadopitys verticillata (Sciadopityaceae). Conserv. Genet. 2014, 15, 1243–1257. [Google Scholar] [CrossRef]
- Chung, S.M.; Staub, J.E.; Lebeda, A.; Paris, H.S. Consensus chloroplast primer analysis: A molecular tool for evolutionary studies in Cucurbitaceae. In Proceedings of the Progress in Cucurbit Genetics and Breeding Research, Olomouc, Czech Republic, 12–17 July 2004. [Google Scholar]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; Mclenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Ao, L.; Chen, J.; Li, W.; Hu, W.; Wei, Z.; Kim, K.; Lee, S.C.; Yang, T.J. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 696. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Birky, C.W., Jr. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1996, 92, 11331–11338. [Google Scholar] [CrossRef]
- Straub, S.C.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Chloroplast genome evolution in Actinidiaceae: clpP Loss, heterogenous divergence and phylogenomic practice. PLoS ONE 2016, 11, e0162324. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M. Next-Generation Organellar Genomics: Potentials and Pitfalls of High-Throughput Technologies for Molecular Evolutionary Studies and Plant Systematics. In Next Generation Sequencing in Plant Systematics; International Association for Plant Taxonomy (IAPT): Bratislava, Slovakia, 2015. [Google Scholar]
- Chen, S.Y.; Zhang, X.Z. Characterization of the complete chloroplast genome of the relict Chinese false tupelo, Camptotheca acuminata. Conserv. Genet. Resour. 2017, 1–4. [Google Scholar] [CrossRef]
- Yang, Z.; Ji, Y. Comparative and phylogenetic analyses of the complete chloroplast genomes of three Arcto-Tertiary relicts: Camptotheca acuminata, Davidia involucrata, and Nyssa sinensis. Front. Plant Sci. 2017, 8, 1536. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, H.; He, Q.; Yang, W.L.; Chen, Z.; Wang, M.; Su, Y.; Ma, T. Characterization of the complete chloroplast genome of Camptotheca acuminata. Conserv. Genet. Resour. 2017, 9, 241–243. [Google Scholar] [CrossRef]
- Puterova, J.; Razumova, O.; Martinek, T.; Alexandrov, O.; Divashuk, M.; Kubat, Z.; Hobza, R.; Karlov, G.; Kejnovsky, E. Satellite DNA and transposable elements in seabuckthorn (Hippophae rhamnoides), a dioecious plant with small Y and large X chromosomes. Genome Biol. Evol. 2017, 9, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.C. An appraisal of the potential for illegitimate recombination in bacterial genomes and its consequences: From duplications to genome reduction. Genome Res. 2003, 13, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Kegel, A.; Martinez, P.; Carter, S.D.; Aström, S.U. Genome wide distribution of illegitimate recombination events in Kluyveromyces lactis. Nucleic Acids Res. 2006, 34, 1633–1645. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dodsworth, S.; Leitch, A.R.; Leitch, I.J. Genome size diversity in angiosperms and its influence on gene space. Curr. Opin. Genet. Dev. 2015, 35, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yonezawa, T.; Zhong, Y.; Hasegawa, M. Episodic evolution and adaptation of chloroplast genomes in ancestral grasses. PLoS ONE 2009, 4, e5297. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gerstein, M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003, 31, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Yamazaki, Y.; Ogihara, Y.; Tsunewaki, K. Whole chloroplast genome comparison of rice, maize, and wheat: Implications for chloroplast gene diversification and phylogeny of cereals. Mol. Biol. Evol. 2002, 19, 2084–2091. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castle, J.C. SNPs occur in regions with less genomic sequence conservation. PLoS ONE 2011, 6, e20660. [Google Scholar] [CrossRef] [PubMed]
- Allegre, M.; Argout, X.; Boccara, M.; Fouet, O.; Roguet, Y.; Bérard, A.; Thévenin, J.M.; Chauveau, A.; Rivallan, R.; Clement, D. Discovery and mapping of a new expressed sequence tag-single nucleotide polymorphism and simple sequence repeat panel for large-scale genetic studies and breeding of Theobroma cacao L. DNA Res. 2011, 19, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, X. Identification of the sex-biased gene expression and putative sex-associated genes in Eucommia ulmoides Oliver using comparative transcriptome analyses. Molecules 2017, 22, 2255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Ma, P.-F.; Li, D.-Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [PubMed]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc. Natl. Acad. Sci. USA 2008, 105, 18424–18429. [Google Scholar] [CrossRef] [PubMed]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [PubMed]
- Bock, D.G.; Kane, N.C.; Ebert, D.P.; Rieseberg, L.H. Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: Neither from Jerusalem nor an artichoke. New Phytol. 2014, 201, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, X.; Zhang, C.; Yin, X.; Liu, S.; Li, X. Development of chloroplast microsatellite markers and analysis of chloroplast diversity in Chinese jujube (Ziziphus jujuba Mill.) and wild jujube (Ziziphus acidojujuba Mill.). PLoS ONE 2015, 10, e0134519. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Jiang, H.; Yan, Z.; Chen, Y.; Zhou, X.; Huang, L.; Lei, Y.; Huang, J.; Yan, L.; Qi, Y. Genetic diversity and population structure of the major peanut (Arachis hypogaea L.) cultivars grown in China by SSR markers. PLoS ONE 2014, 9, e88091. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.D.; Yang, T.; Lin, L.; Lu, L.M.; Li, H.L.; Sun, M.; Liu, B.; Chen, M.; Niu, Y.T.; Ye, J.F. Tree of life for the genera of Chinese vascular plants. J. Syst. Evol. 2016, 54, 277–306. [Google Scholar] [CrossRef]
- Byng, J.W.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Judd, W.S.; Mabberley, D.J.; Sennikov, A.N.; Soltis, D.E.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 105–121. [Google Scholar]
- Stevens, P.F. Angiosperm Phylogeny Website. Version 12, July 2012 (and More or Less Continuously Updated Since). Available online: http://www.mobot.org/MOBOT/research/APweb/ (accessed on 1 March 2018).
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebensmack, J.; Müller, K.F.; Guisingerbellian, M.; Haberle, R.C.; Hansen, A.K. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Beaulieu, J.M.; Donoghue, M.J. An uncorrelated relaxed-clock analysis suggests an earlier origin for flowering plants. Proc. Natl. Acad. Sci. USA 2010, 107, 5897–5902. [Google Scholar] [CrossRef]
- Davis, C.C.; Xi, Z.; Mathews, S. Plastid phylogenomics and green plant phylogeny: Almost full circle but not quite there. BMC Biol. 2014, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Q.; Sun, R.; Kong, H.; Zhang, N.; Ma, H.; Zeng, L.; Zhang, Q.; Sun, R.; Kong, H. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 2014, 5, 4956. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33 (Suppl. 2), W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, L.; Becher, H.; Garcia, S.; Kovarikova, A.; Leitch, I.J.; Leitch, A.R.; Kovarik, A. Astonishing 35S rDNA diversity in the gymnosperm speciesCycas revolutaThunb. Chromosoma 2016, 125, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Y.; Peng, Z.; Sun, H.; Yue, X.; Lou, Y.; Dong, L.; Wang, L.; Gao, Z. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 2015, 5, 8018. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map (SAM) format and SAMtools. Transpl. Proc. 2009, 19, 1653–1654. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–33. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony, version 4.0 b10; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | This Study | KU204775 |
|---|---|---|
| Chloroplast genome size (bp) | 163,586 | 163,341 |
| LSC a length (bp) | 86,764 | 86,592 |
| SSC b length (bp) | 14,166 | 14,149 |
| IRa/IRb c length (bp) | 31,328 | 31,300 |
| Number of genes (unique genes) | 136 (115) | 136 (115) |
| Number of protein-coding genes (unique genes) | 89 (80) | 89 (80) |
| Number of tRNA genes (unique genes) | 39 (31) | 39 (31) |
| Number of rRNA genes (unique genes) | 8 (4) | 8 (4) |
| GC d content (%) | 38.33% | 38.34% |
| Protein-coding regions (%) | 51.91% | 51.99% |
| No. | Size (bp) | Start Position | Location | Type |
|---|---|---|---|---|
| 1 | 56 | 6851 | rps16-trnT(UGU) | insertion |
| 2 | 27 | 7006 | rps16-trnT(UGU) | insertion |
| 3 | 45 | 7196 | rps16-trnT(UGU) | insertion |
| 4 | 13 | 12,693 | ycf3-psaA | insertion |
| 5 | 23 | 12,912 | ycf3-psaA | insertion |
| 6 | 111 | 13,312 | ycf3-psaA | insertion |
| 7 | 12 | 13,471 | ycf3-psaA | insertion |
| 8 | 32 | 24,279 | psbD-trnT(GGU) | insertion |
| 9 | 12 | 26,615 | trnD(GUC)-psbM | insertion |
| 10 | 11 | 51,194 | rps12-rpl20 | insertion |
| 11 | 17 | 52,547 | rps18-rpl33 | insertion |
| 12 | 40 | 57,075 | psbJ-petA | insertion |
| 13 | 18 | 64,506 | accD-trnM(CAU) | insertion |
| 14 | 12 | 73,717 | trnL(UAA) intron | insertion |
| 15 | 14 | 127,486 | ndhG-ndhI | insertion |
| 16 | 16 | 4673 | trnK(UUU)-rps16 | deletion |
| 17 | 44 | 24,700 | trnG(TTU)-trnE(UUC) | deletion |
| 18 | 31 | 41,767 | atpI-atpH | deletion |
| 19 | 44 | 51,109 | rps12-rpl20 | deletion |
| 20 | 90 | 62,865 | accD-trnM(CAU) | deletion |
| Region | Aligned Length (bp) | No. VCs a | Percentage of VCs (%) |
|---|---|---|---|
| infA | 234 | 3 | 1.28 |
| rps18-rpl33 | 343 | 4 | 1.17 |
| rps12 | 369 | 3 | 0.81 |
| rrn5-trnR(ACG) | 266 | 2 | 0.75 |
| ycf15 | 210 | 1 | 0.48 |
| trnI(GAU) | 1062 | 5 | 0.47 |
| petB | 651 | 3 | 0.46 |
| ycf3-psaA | 1502 | 6 | 0.40 |
| trnT(GGU)-atpE | 261 | 1 | 0.38 |
| ndhG | 531 | 2 | 0.38 |
| ycf4 | 546 | 2 | 0.37 |
| trnG(UCC)-psbZ | 280 | 1 | 0.36 |
| rps16 | 1170 | 4 | 0.34 |
| trnA(UGG) | 881 | 3 | 0.34 |
| ndhB-rps7 | 325 | 1 | 0.31 |
| psbK-trnQ(UUG) | 338 | 1 | 0.30 |
| trnI(CAU)-ycf2 | 357 | 1 | 0.28 |
| ycf15-trnL(CAA) | 359 | 1 | 0.28 |
| psbB | 1521 | 4 | 0.27 |
| rpl20 | 384 | 1 | 0.26 |
| No. | SSR Repeat Motif | Length Variation (bp) | Location | Region a |
|---|---|---|---|---|
| 1 | (G) | 10–11 | trnG(UCC)-psbZ | LSC |
| 2 | (A) | 12–15 | rpoC2 | LSC |
| 3 | (A) | 12–13 | ycf1 | IRb |
| 4 | (A) | 13–14 | rpl32-trnL(UAG) | IRa |
| 5 | (T) | 10–14 | psbJ-petA | LSC |
| 6 | (T) | 10–11 | trnG(GCC)-trnS(GCU) | LSC |
| 7 | (T) | 12–13 | ycf1 | IRa |
| 8 | (T) | 14–15 | rpl16-rps3 | LSC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Chen, S.; Zhang, X. Whole-Genome Comparison Reveals Heterogeneous Divergence and Mutation Hotspots in Chloroplast Genome of Eucommia ulmoides Oliver. Int. J. Mol. Sci. 2018, 19, 1037. https://doi.org/10.3390/ijms19041037
Wang W, Chen S, Zhang X. Whole-Genome Comparison Reveals Heterogeneous Divergence and Mutation Hotspots in Chloroplast Genome of Eucommia ulmoides Oliver. International Journal of Molecular Sciences. 2018; 19(4):1037. https://doi.org/10.3390/ijms19041037
Chicago/Turabian StyleWang, Wencai, Siyun Chen, and Xianzhi Zhang. 2018. "Whole-Genome Comparison Reveals Heterogeneous Divergence and Mutation Hotspots in Chloroplast Genome of Eucommia ulmoides Oliver" International Journal of Molecular Sciences 19, no. 4: 1037. https://doi.org/10.3390/ijms19041037
APA StyleWang, W., Chen, S., & Zhang, X. (2018). Whole-Genome Comparison Reveals Heterogeneous Divergence and Mutation Hotspots in Chloroplast Genome of Eucommia ulmoides Oliver. International Journal of Molecular Sciences, 19(4), 1037. https://doi.org/10.3390/ijms19041037