p53 as a Dichotomous Regulator of Liver Disease: The Dose Makes the Medicine

 ,
, 

Abstract
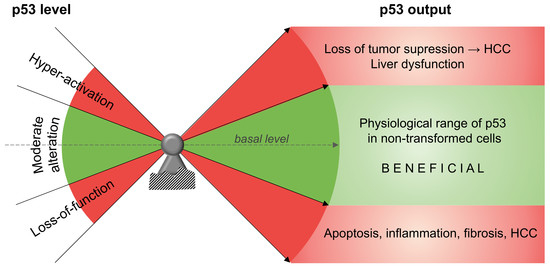
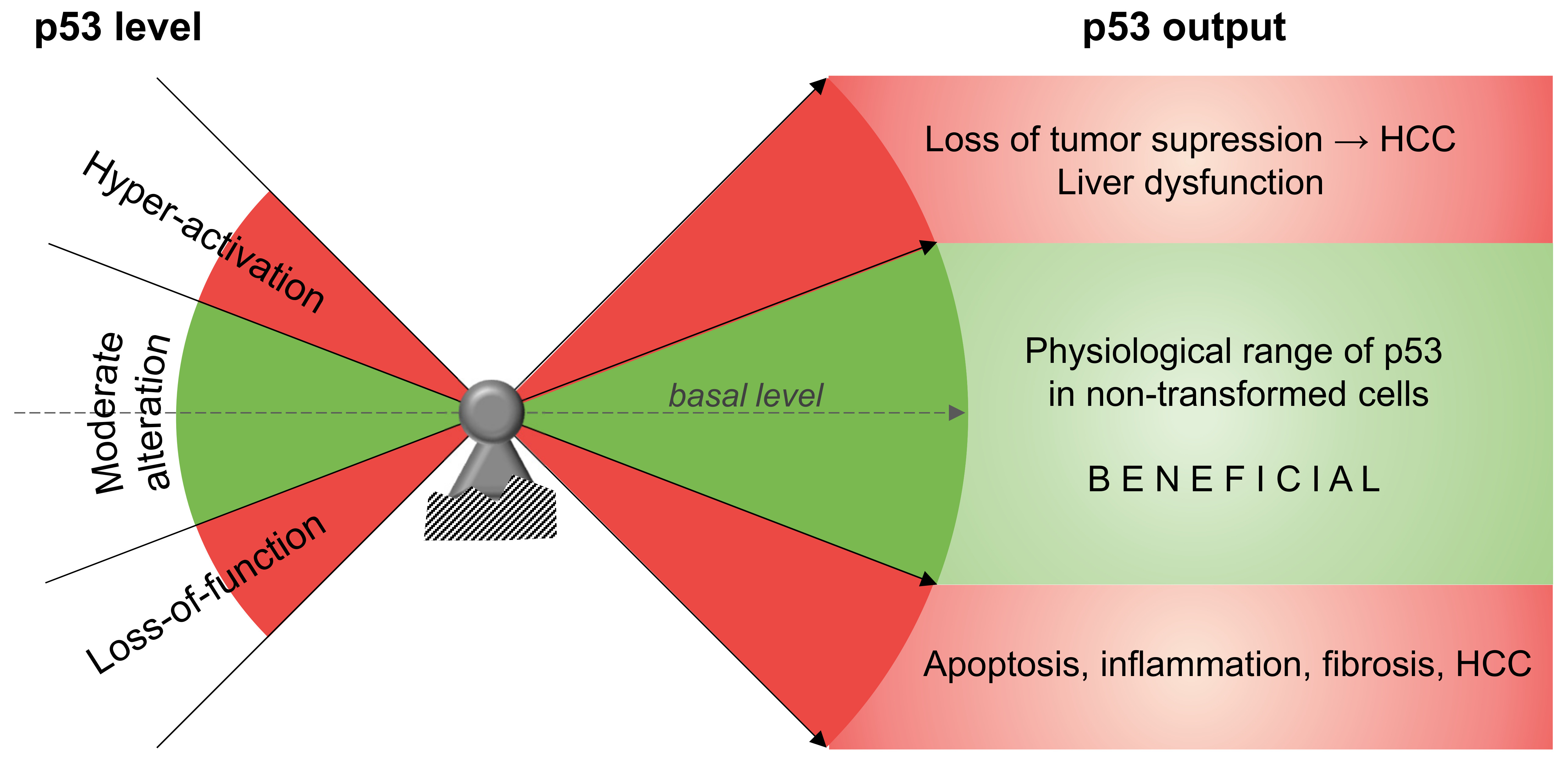{kind=link}
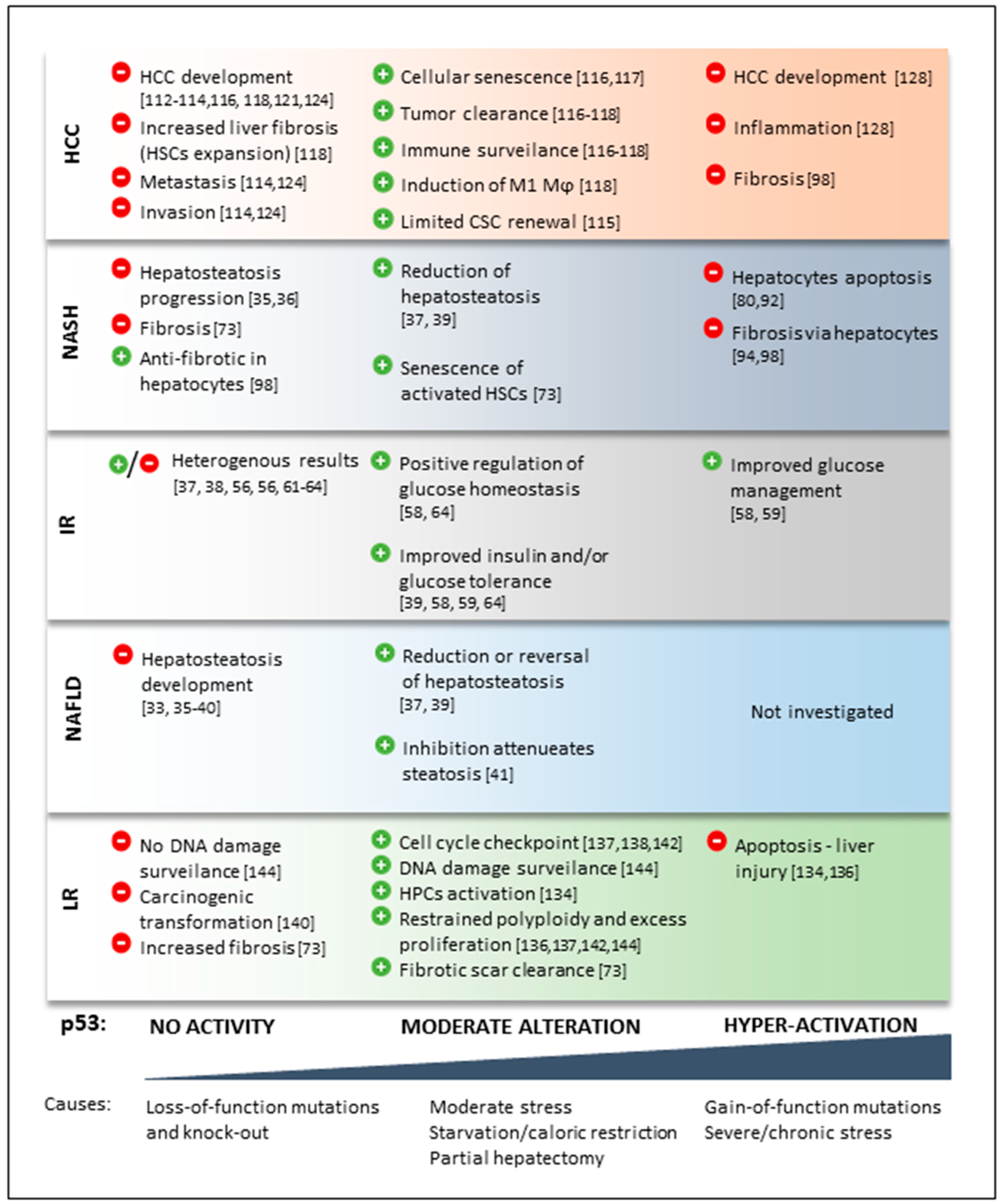{kind=link}
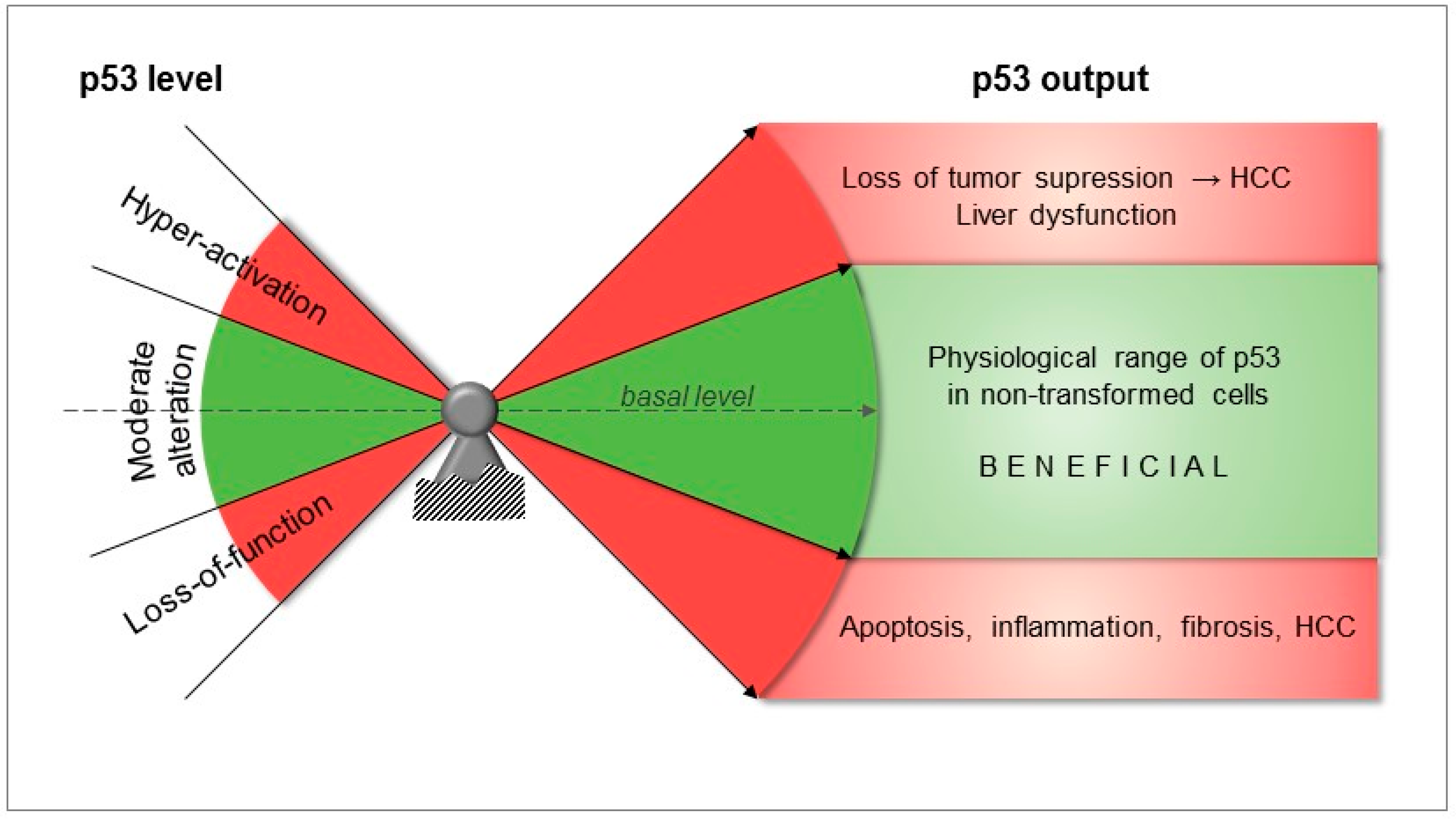{kind=link}
1. Liver Disease in the Age of Metabolic Syndrome
2. p53 in the Development of Non-Alcoholic Fatty Liver Disease
3. p53 in the Regulation of Hepatic Insulin Resistance
4. p53 in the Development of Non-Alcoholic Steatohepatitis
5. Multifunctional Roles of p53 in Hepatocellular Carcinoma
6. p53 in the Regulation of Liver Regeneration
7. Fine-tuning of Intracellular p53 Protein Levels
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brunt, E.M.; Wong, V.W.S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Sun, Z.; Lazar, M.A. Dissociating fatty liver and diabetes. Trends Endocrinol. Metab. 2013, 24, 4–12. [Google Scholar] [CrossRef] [PubMed]
- DeWeerdt, S. Disease progression: Divergent paths. Nature 2017, 551. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C.P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship Between Methylome and Transcriptome in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.Y.L. Drug development: Sprint finish. Nature 2017, 551. [Google Scholar] [CrossRef]
- Younossi, Z.; Henry, L. Contribution of Alcoholic and Nonalcoholic Fatty Liver Disease to the Burden of Liver-Related Morbidity and Mortality. Gastroenterology 2016, 150, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Campos, V.C.; Tappy, L. Physiological handling of dietary fructose-containing sugars: Implications for health. Int. J. Obes. 2016, 40, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Diagnostics: Missing the point. Nature 2017, 551. [Google Scholar] [CrossRef]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Cervello, M.; Bachvarov, D.; Lampiasi, N.; Cusimano, A.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 2012, 11, 2843–2855. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Varley, J.M. Germline TP53 mutations and Li-Fraumeni syndrome. Hum. Mutat. 2003, 21, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.K.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic Regulation by p53 Family Members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Schupp, M.; Chen, F.; Briggs, E.R.; Rao, S.; Pelzmann, H.J.; Pessentheiner, A.R.; Bogner-Strauss, J.G.; Lazar, M.A.; Baldwin, D.; Prokesch, A. Metabolite and transcriptome analysis during fasting suggest a role for the p53-Ddit4 axis in major metabolic tissues. BMC Genom. 2013, 14, 758. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Charni, M.; Rivlin, N.; Molchadsky, A.; Aloni-Grinstein, R.; Rotter, V. P53 in Liver Pathologies—Taking the Good with the Bad. J. Mol. Med. 2014, 92, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Araújo, L.M.; De Oliveira, D.A.; Nunes, D.S. Liver and biliary ultrasonography in diabetic and non-diabetic obese women. Diabetes Metab. 1998, 24, 458–462. [Google Scholar] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Halpern, Z.; Oren, R. Prevalence of primary non-alcoholic fatty liver disease in a population-based study and its association with biochemical and anthropometric measures. Liver Int. 2006, 26, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.F.Y.; Li, A.M.; Chu, W.C.W.; Chan, M.H.M.; Wong, E.M.C.; Liu, E.K.H.; Chan, I.H.S.; Yin, J.; Lam, C.W.K.; Fok, T.F.; et al. Hepatic steatosis in obese Chinese children. Int. J. Obes. 2004, 28, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Panasiuk, A. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 2006, 12, 6198. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Sekiya, M.; Najima, Y.; Okazaki, S.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Inoue, N.; et al. p53 Involvement in the Pathogenesis of Fatty Liver Disease. J. Biol. Chem. 2004, 279, 20571–20575. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Gao, X.; Mei, Y.; Wu, M. A new role of p53 in regulating lipid metabolism. J. Mol. Cell Biol. 2013, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Porteiro, B.; Fondevila, M.F.; Delgado, T.C.; Iglesias, C.; Imbernon, M.; Iruzubieta, P.; Crespo, J.; Zabala-Letona, A.; Fernø, J.; González-Terán, B.; et al. Hepatic p63 regulates steatosis via IKKβ/ER stress. Nat. Commun. 2017, 8, 15111. [Google Scholar] [CrossRef] [PubMed]
- Prokesch, A.; Graef, F.A.; Madl, T.; Kahlhofer, J.; Heidenreich, S.; Schumann, A.; Moyschewitz, E.; Pristoynik, P.; Blaschitz, A.; Knauer, M.; et al. Liver p53 is stabilized upon starvation and required for amino acid catabolism and gluconeogenesis. FASEB J. 2017, 31, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Porteiro, B.; Fondevila, M.F.; Buque, X.; Gonzalez-Rellan, M.J.; Fernandez, U.; Mora, A.; Beiroa, D.; Senra, A.; Gallego, R.; Fernø, J.; et al. Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Mol. Metab. 2018, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, Y.; Jin, A.; Tikunov, A.P.; Zhou, L.; Tollini, L.A.; Leslie, P.; Kim, T.-H.; Li, L.O.; Coleman, R.A.; et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, E2414–E2422. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Neitemeier, S.; Ganjam, G.K.; Diemert, S.; Culmsee, C. Pifithrin-α provides neuroprotective effects at the level of mitochondria independently of p53 inhibition. Apoptosis 2014, 19, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.; Kurauchi, K.; Tomizawa, A.; Yomogida, S.; Ishikawa, M. Pifithrin-alpha has a p53-independent cytoprotective effect on docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma HepG2 cells. Toxicol. Lett. 2015, 232, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Rotter, V. Regulation of lipid metabolism by p53—Fighting two villains with one sword. Trends Endocrinol. Metab. 2012, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Flöter, J.; Kaymak, I.; Schulze, A. Regulation of Metabolic Activity by p53. Metabolites 2017, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-J.; Yu, G.; Jiang, L.; Li, T.; Lin, Q.; Tang, Y.; Gu, W. p53-dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle 2013, 12, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Leavens, K.F.; Hunter, R.W.; Koren, S.; Von Wilamowitz-Moellendorff, A.; Lu, M.; Satapati, S.; Chu, Q.; Sakamoto, K.; Burgess, S.C.; et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 2013, 18, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Investig. 2002, 109, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Moitra, J.; Mason, M.M.; Olive, M.; Krylov, D.; Gavrilova, O.; Marcus-Samuels, B.; Feigenbaum, L.; Lee, E.; Aoyama, T.; Eckhaus, M.; et al. Life without white fat: A transgenic mouse. Genes Dev. 1998, 12, 3168–3181. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Ayala, J.E.; Bracy, D.P.; James, F.D.; Burmeister, M.A.; Wasserman, D.H.; Drucker, D.J. Glucagon-Like Peptide-1 Receptor Knockout Mice Are Protected from High-Fat Diet-Induced Insulin Resistance. Endocrinology 2010, 151, 4678–4687. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.-P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Armata, H.L.; Golebiowski, D.; Jung, D.Y.; Ko, H.J.; Kim, J.K.; Sluss, H.K. Requirement of the ATM/p53 Tumor Suppressor Pathway for Glucose Homeostasis. Mol. Cell. Biol. 2010, 30, 5787–5794. [Google Scholar] [CrossRef] [PubMed]
- García-Cao, I.; García-Cao, M.; Martín-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.-C.; Blasco, M.A.; Serrano, M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef] [PubMed]
- Franck, D.; Tracy, L.; Armata, H.L.; Delaney, C.L.; Jung, D.Y.; Ko, H.J.; Ong, H.; Kim, J.K.; Scrable, H.; Sluss, H.K. Glucose Tolerance in Mice is Linked to the Dose of the p53 Transactivation Domain. Endocr. Res. 2013, 38, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Duan, W.; Lee, W.-N.P.; Zhang, Y.; Xiang, F.; Liu, Q.; Go, V.L.W.; Xiao, G.G. Overexpression of p53 Improves Blood Glucose Control in an Insulin Resistant Diabetic Mouse Model. Pancreas 2016, 45, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Lang, C.H.; Villegas, K.A.; Tong, M.; Mark, N.M.; de la Monte, S.M.; Wands, J.R. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J. Hepatol. 2011, 54, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tu, B.; Wang, H.; Cao, Z.; Tang, M.; Zhang, C.; Gu, B.; Li, Z.; Wang, L.; Yang, Y.; et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 2014, 111, 10684–10689. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, A.J.; Rogoff, D.; White, P.C. P53 and cellular glucose uptake. Endocr. Res. 2013, 38, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Leu, J.I.-J.; Basu, S.; Khaku, S.; Anokye-Danso, F.; Liu, Q.; George, D.L.; Ahima, R.S.; Murphy, M.E. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep. 2016, 14, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Basu, S.; Murphy, M.E. A link between TP53 polymorphisms and metabolism. Mol. Cell. Oncol. 2016, 3, e1173769. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, K.S.; Grarup, N.; Justesen, J.M.; Harder, M.N.; Witte, D.R.; Jørgensen, T.; Sandbæk, A.; Lauritzen, T.; Madsbad, S.; Hansen, T.; et al. Studies of the association of Arg72Pro of tumor suppressor protein p53 with type 2 diabetes in a combined analysis of 55,521 Europeans. PLoS ONE 2011, 6, e15813. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology 2009, 49, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Yizhak, K.; Madar, S.; Goldfinger, N.; Ruppin, E.; Rotter, V. p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab. 2013, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Guo, J.; Cao, Y.; Wang, S.; Pang, C.; Li, M.; Dou, L.; Man, Y.; Huang, X.; Shen, T.; et al. MicroRNA-20a-5p contributes to hepatic glycogen synthesis through targeting p63 to regulate p53 and PTEN expression. J. Cell. Mol. Med. 2016, 20, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Strycharz, J.; Drzewoski, J.; Szemraj, J.; Sliwinska, A. Is p53 Involved in Tissue-Specific Insulin Resistance Formation? Oxid. Med. Cell. Longev. 2017, 2017, 9270549. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic Fatty Liver Disease: Pathology and Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z.; Hou, J.Y.; Zhang, R.H.; Yeh, M.M.; Williams, J.; Dela Peňa, A.; Francisco, R.; Osvath, S.R.; Brooling, J.; et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 2009, 24, 443–452. [Google Scholar] [CrossRef] [PubMed]
- García-Galiano, D.; Sánchez-Garrido, M.A.; Espejo, I.; Montero, J.L.; Costán, G.; Marchal, T.; Membrives, A.; Gallardo-Valverde, J.M.; Muñoz-Castañeda, J.R.; Arévalo, E.; et al. IL-6 and IGF-1 are Independent Prognostic Factors of Liver Steatosis and Non-Alcoholic Steatohepatitis in Morbidly Obese Patients. Obes. Surg. 2007, 17, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yonei, Y.; Tanaka, S.; Mori, K.; Kanemasa, K.; Imai, S.; Taketani, H.; Hara, T.; Seko, Y.; Ishiba, H.; et al. Lower levels of insulin-like growth factor-1 standard deviation score are associated with histological severity of non-alcoholic fatty liver disease. Hepatol. Res. 2015, 45, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; McDonald, E.R.; Meng, R.D.; Kao, G.; Muschel, R.; Yen, T.; El-Deiry, W.S. Induction of the TRAIL receptor KILLER/DR5 in p53-dependent apoptosis but not growth arrest. Oncogene 1999, 18, 6411–6418. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, K.; Krigsfeld, G.; Wang, W.; Xu, J.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. TNFSF10 (TRAIL), a p53 target gene that mediates p53-dependent cell death. Cancer Biol. Ther. 2008, 7, 2034–2038. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lowe, S.W.; Hannon, G.J. microRNAs join the p53 network—Another piece in the tumour-suppression puzzle. Nat. Rev. Cancer 2007, 7, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Starlard-Davenport, A.; Tryndyak, V.P.; Han, T.; Ross, S.A.; Rusyn, I.; Beland, F.A. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab. Investig. 2010, 90, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Latendresse, J.R.; Montgomery, B.; Ross, S.A.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Plasma microRNAs are sensitive indicators of inter-strain differences in the severity of liver injury induced in mice by a choline- and folate-deficient diet. Toxicol. Appl. Pharmacol. 2012, 262, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.E.; Ferreira, D.M.S.; Afonso, M.B.; Borralho, P.M.; MacHado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.P. MiR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.M.S.; Afonso, M.B.; Rodrigues, P.M.; Simao, A.L.; Pereira, D.M.; Borralho, P.M.; Rodrigues, C.M.P.; Castro, R.E. c-Jun N-Terminal Kinase 1/c-Jun Activation of the p53/MicroRNA 34a/Sirtuin 1 Pathway Contributes to Apoptosis Induced by Deoxycholic Acid in Rat Liver. Mol. Cell. Biol. 2014, 34, 1100–1120. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.F.; Ji, F.J.; Zang, H.L.; Cao, H. Activation of the miR-34a/SIRT1/p53 signaling pathway contributes to the progress of liver fibrosis via inducing apoptosis in hepatocytes but not in HSCs. PLoS ONE 2016, 11, e0158657. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Park, J.; Wang, S.; Kim, J.; Lee, H.H.; Seo, Y.S.; Jung, Y. MicroRNA expression profiling in CCl4-induced liver fibrosis of mus musculus. Int. J. Mol. Sci. 2016, 17, 961. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Investig. 2011, 121, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Gaddikeri, S.; McNeeley, M.F.; Wang, C.L.; Bhargava, P.; Dighe, M.K.; Yeh, M.M.C.; Dubinsky, T.J.; Kolokythas, O.; Lalwani, N. Hepatocellular carcinoma in the noncirrhotic liver. Am. J. Roentgenol. 2014, 203, W34–W47. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, H. Multiple interactive factors in hepatocarcinogenesis. Cancer Lett. 2014, 346, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xie, L.; Yang, W.S.; Zhang, W.; Gao, S.; Wang, J.; Xiang, Y.B. Risk factors of Hepatocellular Carcinoma—Current Status and Perspectives. Asian Pac. J. Cancer Prev. J Cancer Prev. 2012, 13, 743–752. [Google Scholar] [CrossRef]
- Scalera, A.; Tarantino, G. Could metabolic syndrome lead to hepatocarcinoma via non-alcoholic fatty liver disease? World J. Gastroenterol. 2014, 20, 9217–9228. [Google Scholar] [CrossRef] [PubMed]
- Gillet, R.; Grimber, G.; Bennoun, M.; Caron de Fromentel, C.; Briand, P.; Joulin, V. The consequence of p53 overexpression for liver tumor development and the response of transformed murine hepatocytes to genotoxic agents. Oncogene 2000, 19, 3498–3507. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graur, F.; Furcea, L.; Mois, E.; Biliuta, A.; Rozs, A.T.; Negrean, V.; Hajjar, N. Al Analysis of p53 protein expression in hepatocellular carcinoma. J. Gastrointestig. Liver Dis. 2016, 25, 345–349. [Google Scholar] [CrossRef]
- Liu, J.; Li, W.; Deng, M.; Liu, D.; Ma, Q.; Feng, X. Immunohistochemical determination of p53 protein overexpression for predicting p53 gene mutations in hepatocellular carcinoma: A meta-analysis. PLoS ONE 2016, 11, e0159636. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, Q.; Xue, J. TP53 immunohistochemical expression is associated with the poor outcome for hepatocellular carcinoma: Evidence from a meta-analysis. Tumor Biol. 2014, 35, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.-S.; La, Z.; Yang, L.; He, Q.; Li, P. P53 Gene in Treatment of Hepatic Carcinoma: Status Quo. World J. Gastroenterol. 2007, 13, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Minouchi, K.; Kaneko, S.; Kobayashi, K. Mutation of p53 gene in regenerative nodules in cirrhotic liver. J. Hepatol. 2002, 37, 231–239. [Google Scholar] [CrossRef]
- Katz, S.F.; Lechel, A.; Obenauf, A.C.; Begus-Nahrmann, Y.; Kraus, J.M.; Hoffmann, E.M.; Duda, J.; Eshraghi, P.; Hartmann, D.; Liss, B.; et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology 2012, 142, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. P53-dependent nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Klimstra, D.S.; Mongeau, M.E.; Tatem, J.L.; Boyartchuk, V.; Lewis, B.C. Loss of p53 and Ink4a/Arf cooperate in a cell autonomous fashion to induce metastasis of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 7589–7596. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Dhar, D. Liver carcinogenesis: From naughty chemicals to soothing fat and the surprising role of NRF2. Carcinogenesis 2016, 37, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Kim, D.Y.; Cho, K.J.; Ju, H.-L.; Kim, D.Y.; Ahn, S.H.; Han, K.-H.; Ro, S.W. Development of a transgenic mouse model of hepatocellular carcinoma with a liver fibrosis background. BMC Gastroenterol. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.M.; Frandsen, J.L.; Kirchhof, N.; McIvor, R.S.; Largaespada, D.A. Somatic integration of an oncogene-harboring Sleeping Beauty transposon models liver tumor development in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 17059–17064. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Calvisi, D.F. Hydrodynamic Transfection for Generation of Novel Mouse Models for Liver Cancer Research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.C.; Klimstra, D.S.; Socci, N.D.; Xu, S.; Koutcher, J.A.; Varmus, H.E. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol. Cell. Biol. 2005, 25, 1228–1237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.; Bennoun, M.; Allemand, I.; Molina, T.; Grimber, G.; Daudet-Monsac, M.; Abelanet, R.; Briand, P. Time-course development of differentiated hepatocarcinoma and lung metastasis in transgenic mice. J. Hepatol. 1991, 13, 227–239. [Google Scholar] [CrossRef]
- Allemand, I.; Grimber, G.; Kornprobst, M.; Bennoun, M.; Molina, T.; Briand, P.; Joulin, V. Compensatory apoptosis in response to SV40 large T antigen expression in the liver. Oncogene 1995, 11, 2583–2590. [Google Scholar] [PubMed]
- Yan, H.X.; Wu, H.P.; Zhang, H.L.; Ashton, C.; Tong, C.; Wu, H.; Qian, Q.J.; Wang, H.Y.; Ying, Q.L. P53 promotes inflammation-associated hepatocarcinogenesis by inducing HMGB1 release. J. Hepatol. 2013, 59, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, S.; Hamid, S.; Siddiqui, A.; Hussain, S.; Pervez, S.; Alexander, G. Altered expression of cell cycle and apoptotic proteins in chronic hepatitis C virus infection. BMC Microbiol. 2008, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Elmore, L.W.; Hancock, A.R.; Chang, S.-F.; Wang, X.W.; Chang, S.; Callahan, C.P.; Geller, D.A.; Will, H.; Harris, C.C. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 14707–14712. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Iakova, P.; Jiang, Y.; Lewis, K.; Sullivan, E.; Jawanmardi, N.; Donehower, L.; Timchenko, L.; Timchenko, N.A. Transcriptional and translational regulation of C/EBPβ-HDAC1 protein complexes controls different levels of p53, SIRT1, and PGC1α proteins at the early and late stages of liver cancer. J. Biol. Chem. 2013, 288, 14451–14462. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-renewing diploid Axin2 + cells fuel homeostatic renewal of the liver. Nature 2015, 524, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Alison, M.R.; Lin, W.R. Diverse routes to liver regeneration. J. Pathol. 2016, 238, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity Europe PMC Funders Group. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, L.; He, Z.; Li, G.; Liu, J.; Song, Z.; Jin, H.; Rudolph, K.L.; Yang, H.; Mao, Y.; et al. Inhibition of wild-type p53-induced phosphatase 1 promotes liver regeneration in mice by direct activation of mammalian target of rapamycin. Hepatology 2015, 61, 2030–2041. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Chen, P.; Tan, H.; Zeng, H.; Jiang, Y.; Wang, Y.; Wang, Y.; Hou, X.; Bi, H.; Huang, M. Dynamic and coordinated regulation of KEAP1-NRF2-ARE and p53/p21 signaling pathways is associated with acetaminophen injury responsive liver regeneration. Drug Metab. Dispos. 2014, 42, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Kurinna, S.; Stratton, S.A.; Tsai, W.-W.; Akdemir, K.C.; Gu, W.; Singh, P.; Goode, T.; Darlington, G.J.; Barton, M.C. Direct activation of forkhead box O3 by tumor suppressors p53 and p73 is disrupted during liver regeneration in mice. Hepatology 2010, 52, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Stepniak, E.; Ricci, R.; Eferl, R.; Sumara, G.; Sumara, I.; Rath, M.; Hui, L.; Wagner, E.F. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006, 20, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Charni, M.; Aloni-Grinstein, R.; Molchadsky, A.; Rotter, V. P53 on the crossroad between regeneration and cancer. Cell Death Differ. 2017, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Dumble, M.L.; Croager, E.J.; Yeoh, G.C.T.; Quail, E.A. Generation and characterization of p53 null transformed hepatic progenitor cells: Oval cells give rise to hepatocellular carcinoma. Carcinogenesis 2002, 23, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-C.; Chu, H.-Y.; Yang, C.-W.; Chou, C.-K.; Tsai, T.-F. Aurora-A Overexpression in Mouse Liver Causes p53-Dependent Premitotic Arrest during Liver Regeneration. Mol. Cancer Res. 2009, 7, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Iversen, P.L. Antisense oligonucleotides targeted to the p53 gene modulate liver regeneration in vivo. Drug Metab. Dispos. 2000, 28, 131–138. [Google Scholar] [PubMed]
- Wang, M.-J.; Chen, F.; Lau, J.T.Y.; Hu, Y.-P. Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis. 2017, 8, e2805. [Google Scholar] [CrossRef] [PubMed]
- Kurinna, S.; Stratton, S.A.; Coban, Z.; Schumacher, J.M.; Grompe, M.; Duncan, A.W.; Barton, M.C. P53 Regulates a Mitotic Transcription Program and Determines Ploidy in Normal Mouse Liver. Hepatology 2013, 57, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Lukin, D.J.; Carvajal, L.A.; Liu, W.; Resnick-Silverman, L.; Manfredi, J.J. p53 Promotes Cell Survival due to the Reversibility of Its Cell-Cycle Checkpoints. Mol. Cancer Res. 2015, 13, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Lin, D.C.C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-X.; Wu, H.-P.; Zhang, H.-L.; Ashton, C.; Tong, C.; Wu, J.; Qian, Q.-J.; Wang, H.-Y.; Ying, Q.-L. DNA damage-induced sustained p53 activation contributes to inflammation-associated hepatocarcinogenesis in rats. Oncogene 2013, 32, 4565–4571. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, Y.; Guan, D.; Li, J.; Yin, H.; Pan, Y.; Xie, D.; Chen, Y. Critical Roles of p53 in Epithelial-Mesenchymal Transition and Metastasis of Hepatocellular Carcinoma Cells. PLoS ONE 2013, 8, e72846. [Google Scholar] [CrossRef] [PubMed]
- Sane, S.; Rezvani, K. Essential Roles of E3 Ubiquitin Ligases in p53 Regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Bouchier-Hayes, L.; Kuwana, T.; Newmeyer, D.D.; Green, D.R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005, 309, 1732–1735. [Google Scholar] [CrossRef] [PubMed]
- Lessel, D.; Wu, D.; Trujillo, C.; Ramezani, T.; Lessel, I.; Alwasiyah, M.K.; Saha, B.; Hisama, F.M.; Rading, K.; Goebel, I.; et al. Dysfunction of the MDM2/p53 axis is linked to premature aging. J. Clin. Investig. 2017, 127, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Sluss, H.K.; Armata, H.; Gallant, J.; Jones, S.N. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 2004, 24, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Liu, Q.; Murphy, M.E. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol. Ther. 2017, 18, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. Roles for MicroRNAs in Conferring Robustness to Biological Processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luo, L.; Chen, W.; Xu, H.X.; Chen, F.; Chen, L.Z.; Zeng, W.T.; Chen, J.S.; Huang, X.H. MicroRNA-24 increases hepatocellular carcinoma cell metastasis and invasion by targeting p53: MiR-24 targeted p53. Biomed. Pharmacother. 2016, 84, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, J.; Deng, H.; Wan, H.; Liu, M.; Wang, J.; Li, S.; Li, X.; Tang, H. miR-1228 promotes the proliferation and metastasis of hepatoma cells through a p53 forward feedback loop. Br. J. Cancer 2015, 112, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Simerzin, A.; Zorde-Khvalevsky, E.; Rivkin, M.; Adar, R.; Zucman-Rossi, J.; Couchy, G.; Roskams, T.; Govaere, O.; Oren, M.; Giladi, H.; et al. The liver-specific microRNA-122*, the complementary strand of microRNA-122, acts as a tumor suppressor by modulating the p53/mouse double minute 2 homolog circuitry. Hepatology 2016, 64, 1623–1636. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krstic, J.; Galhuber, M.; Schulz, T.J.; Schupp, M.; Prokesch, A. p53 as a Dichotomous Regulator of Liver Disease: The Dose Makes the Medicine. Int. J. Mol. Sci. 2018, 19, 921. https://doi.org/10.3390/ijms19030921
Krstic J, Galhuber M, Schulz TJ, Schupp M, Prokesch A. p53 as a Dichotomous Regulator of Liver Disease: The Dose Makes the Medicine. International Journal of Molecular Sciences. 2018; 19(3):921. https://doi.org/10.3390/ijms19030921
Chicago/Turabian StyleKrstic, Jelena, Markus Galhuber, Tim J. Schulz, Michael Schupp, and Andreas Prokesch. 2018. "p53 as a Dichotomous Regulator of Liver Disease: The Dose Makes the Medicine" International Journal of Molecular Sciences 19, no. 3: 921. https://doi.org/10.3390/ijms19030921
APA StyleKrstic, J., Galhuber, M., Schulz, T. J., Schupp, M., & Prokesch, A. (2018). p53 as a Dichotomous Regulator of Liver Disease: The Dose Makes the Medicine. International Journal of Molecular Sciences, 19(3), 921. https://doi.org/10.3390/ijms19030921