CpG ODN1826 as a Promising Mucin1-Maltose-Binding Protein Vaccine Adjuvant Induced DC Maturation and Enhanced Antitumor Immunity

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
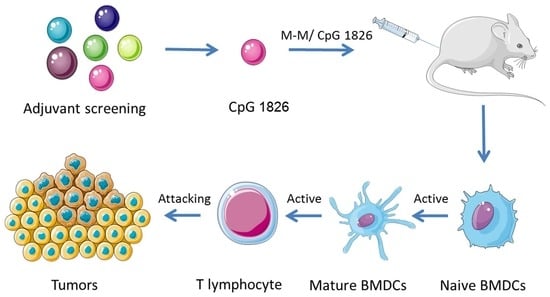
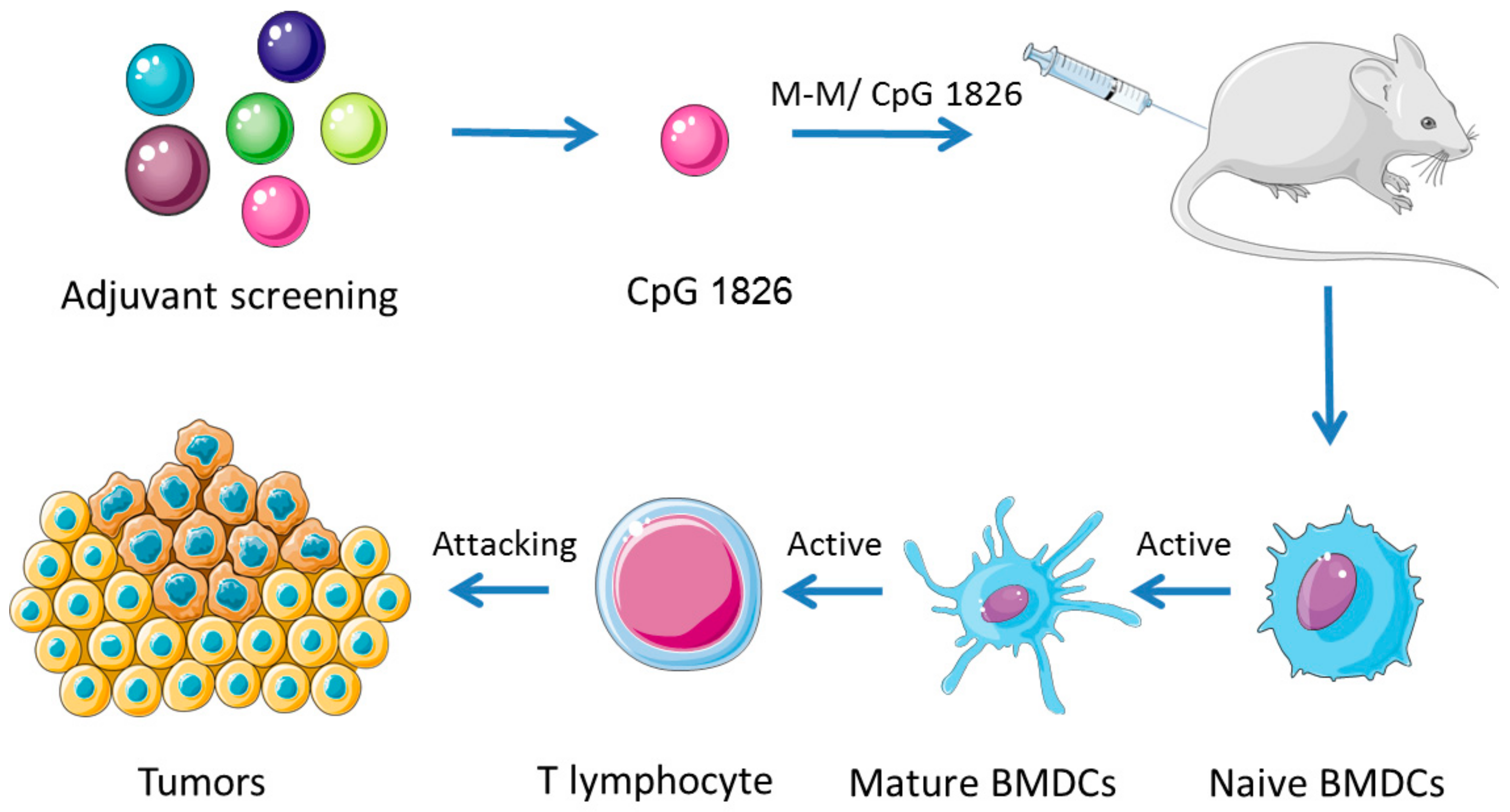
1. Introduction
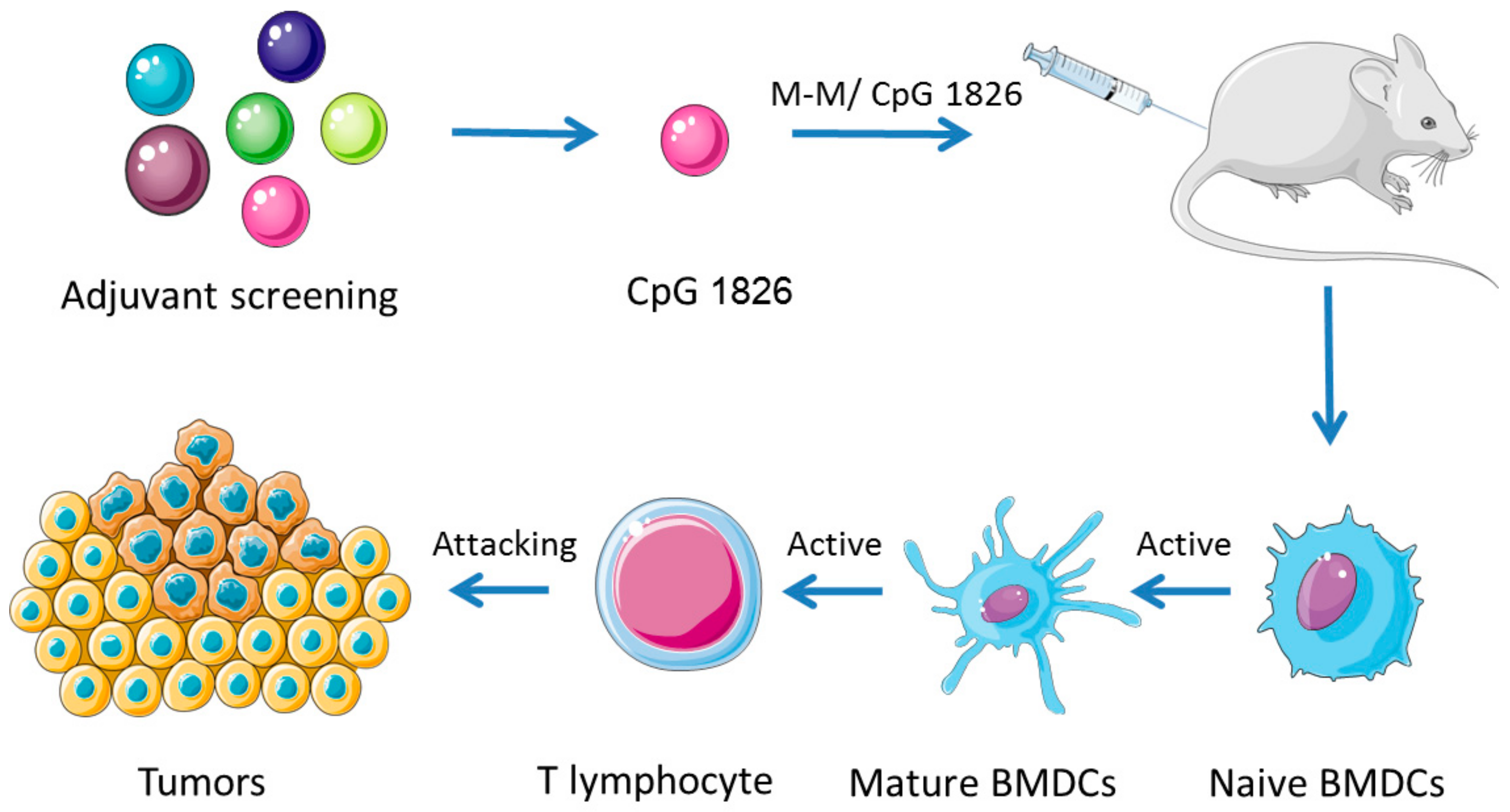
2. Results
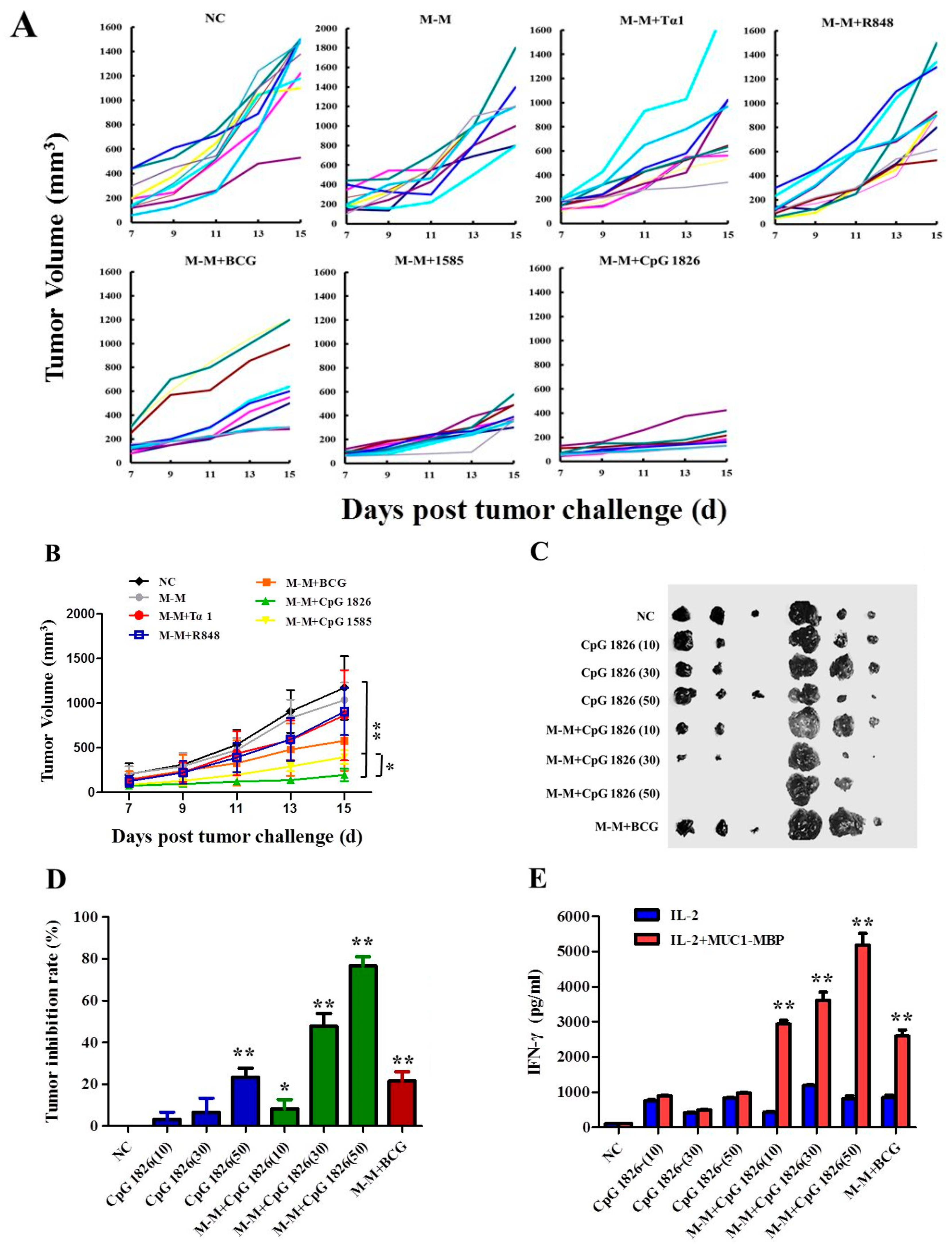
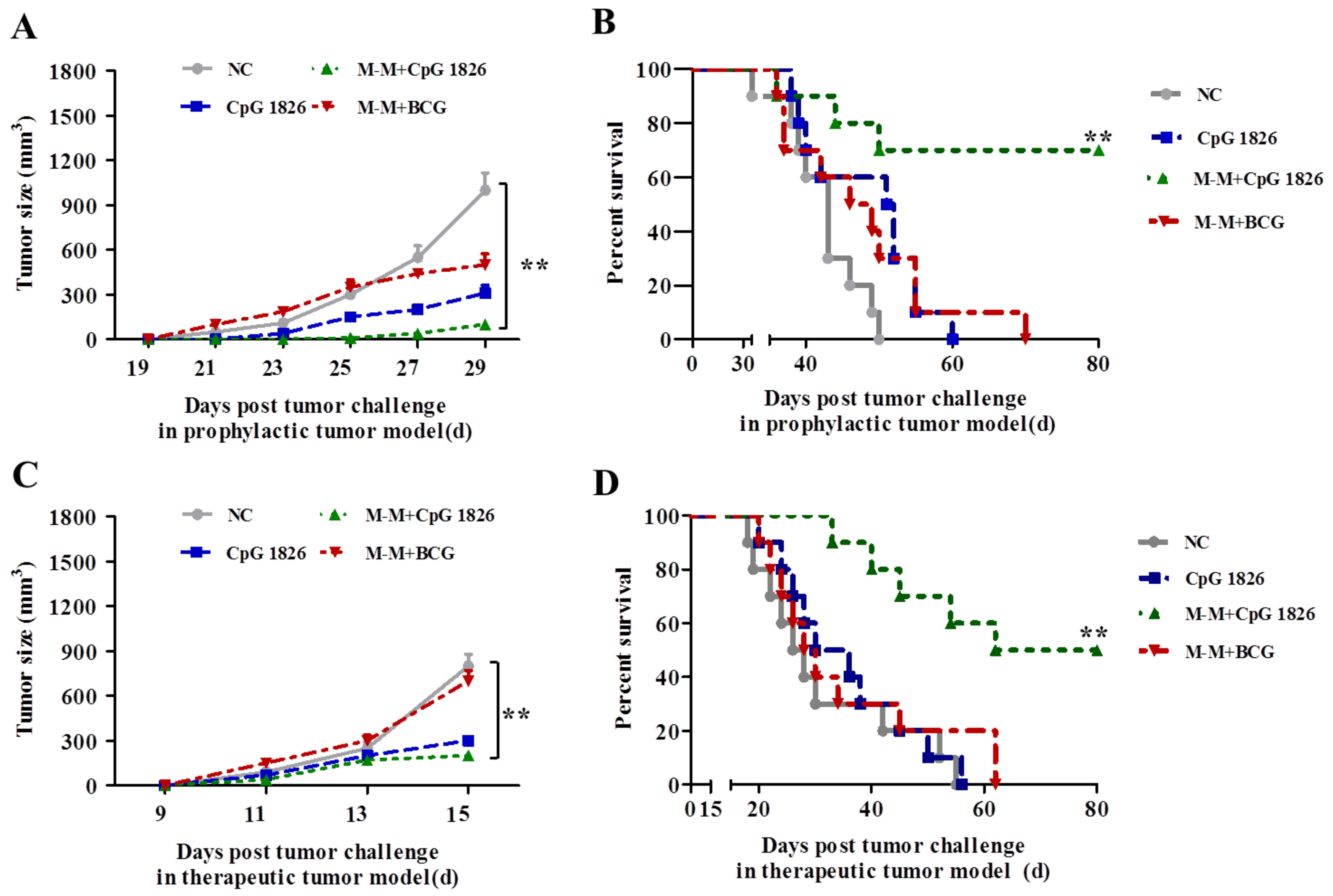
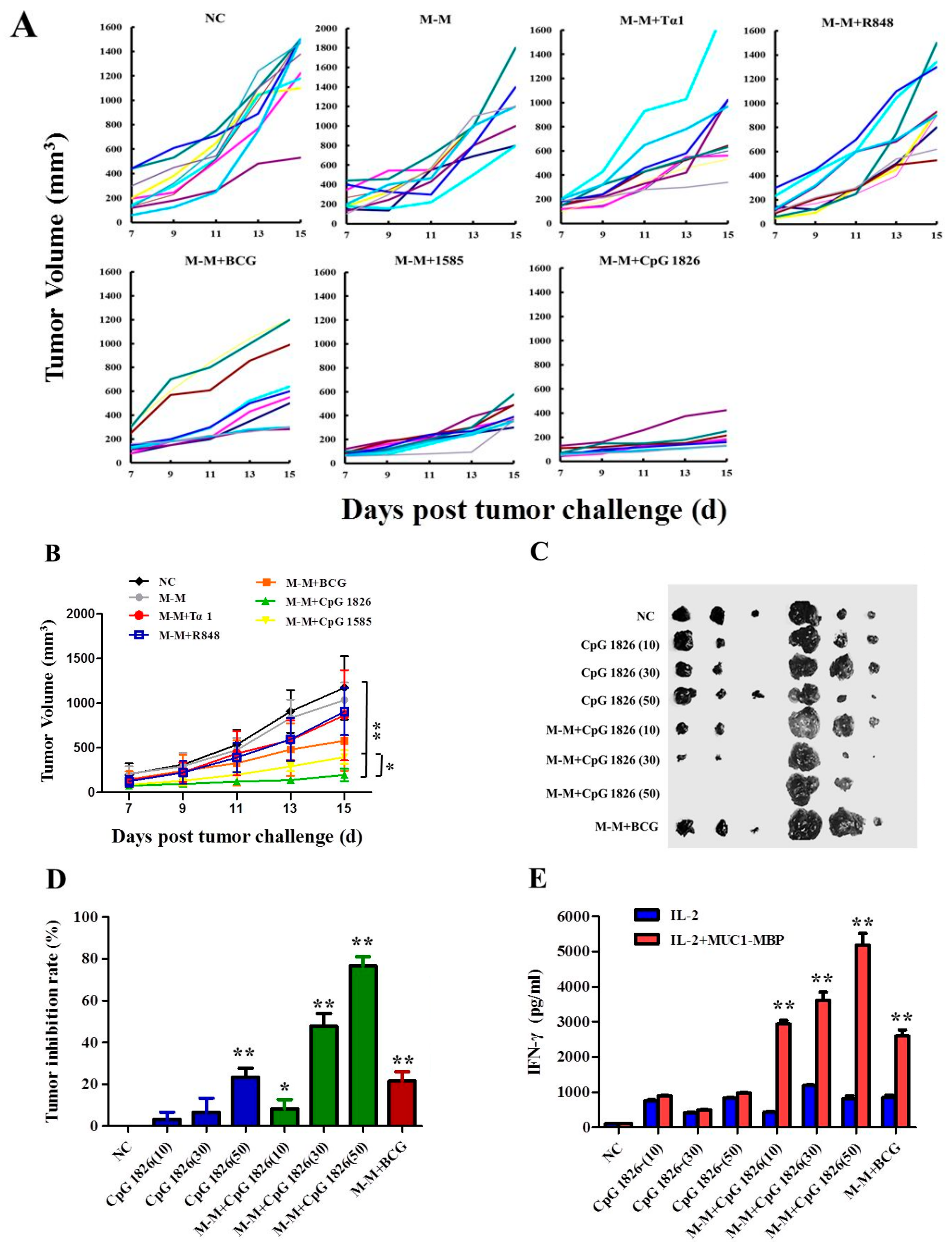
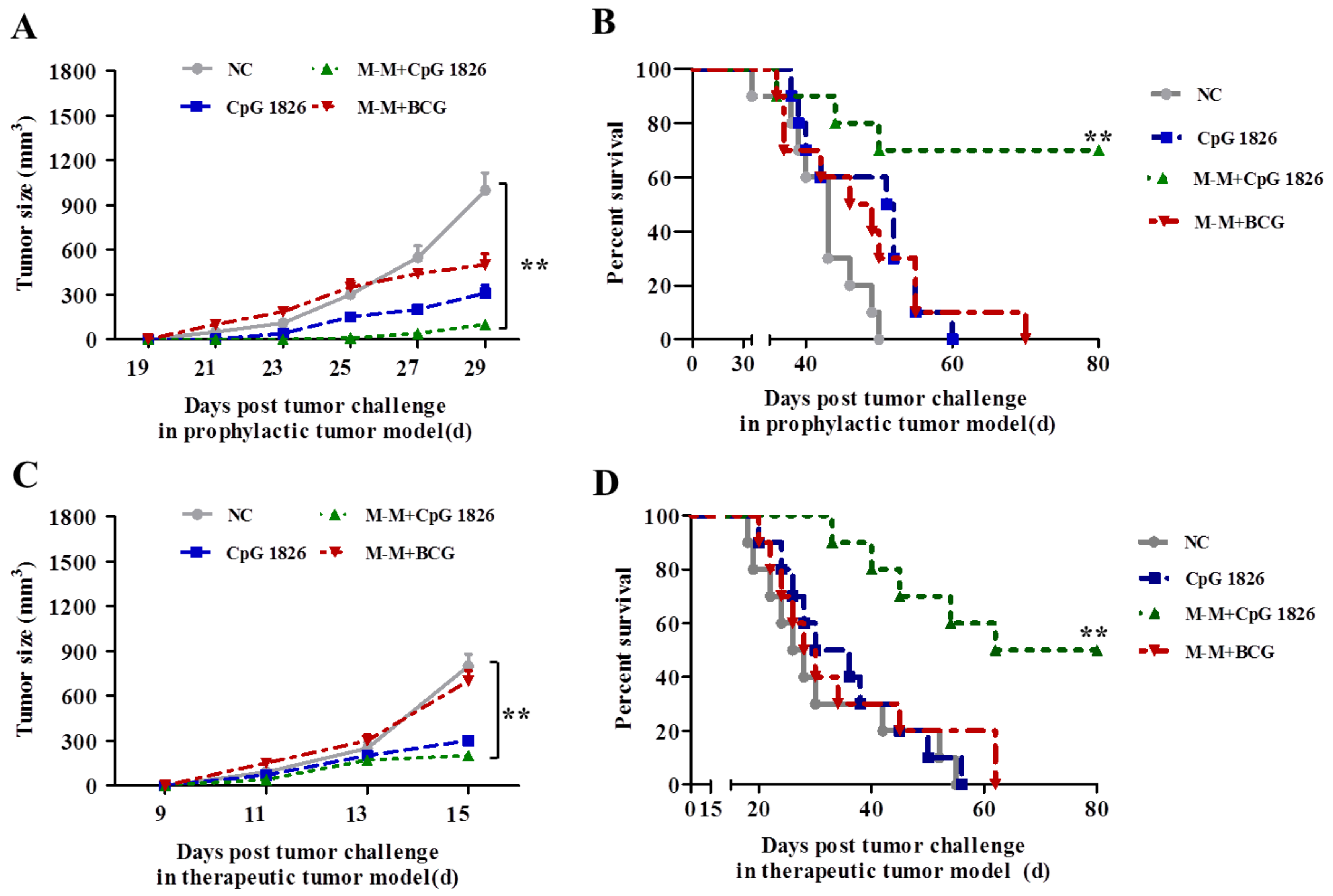
2.1. Combination of M-M and CpG 1826 Inhibited B16-MUC1 Growth in Mice
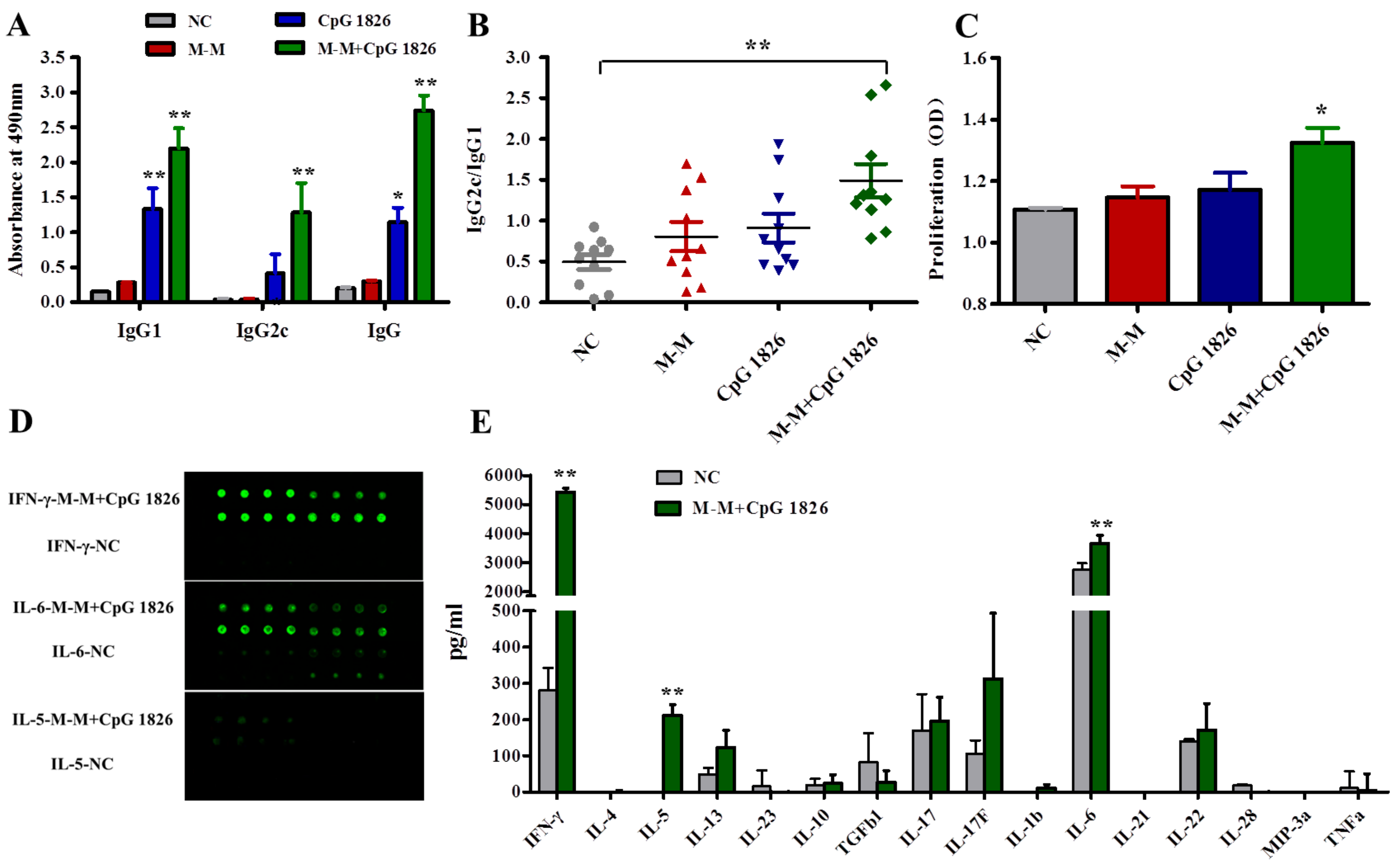
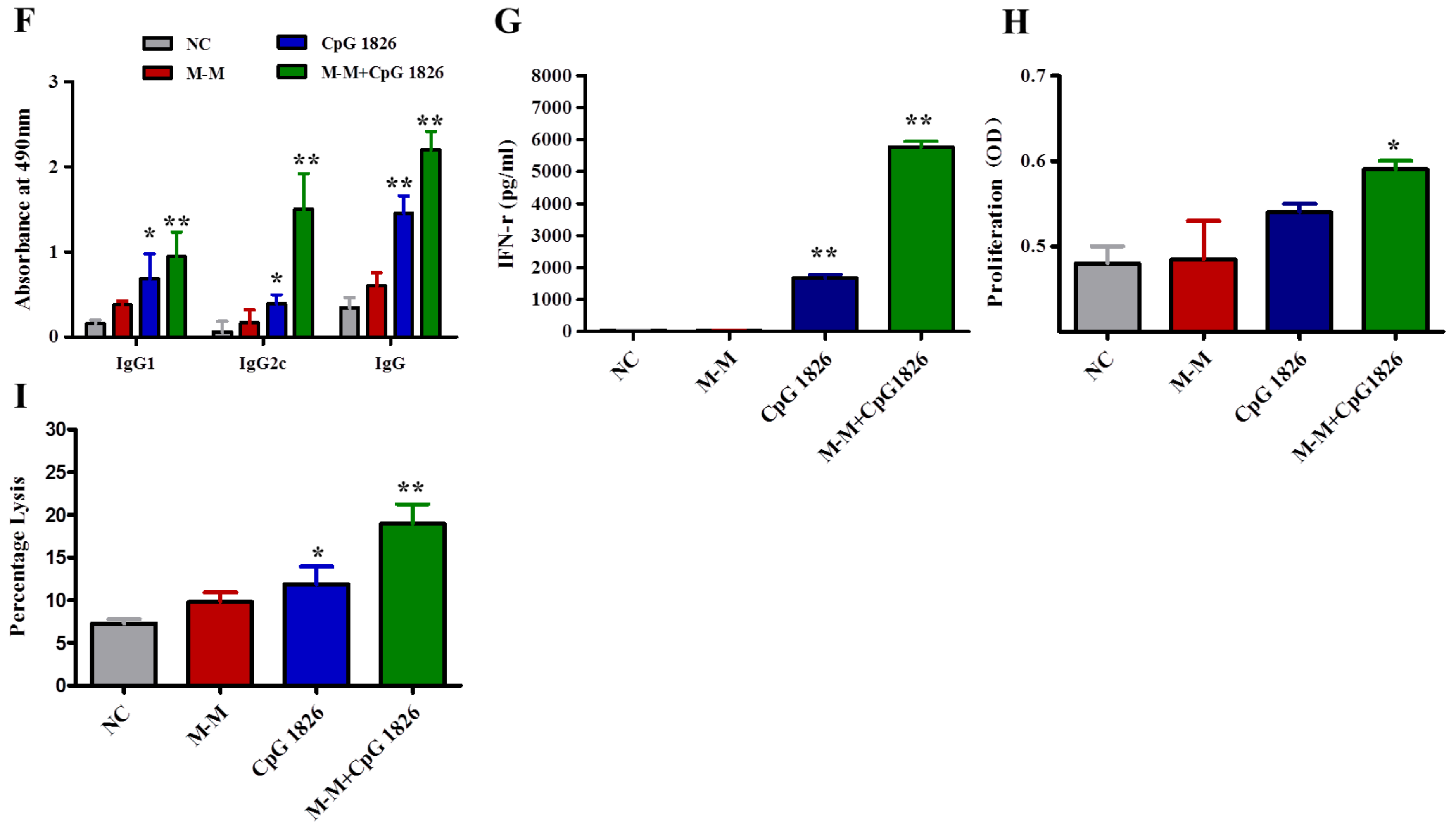
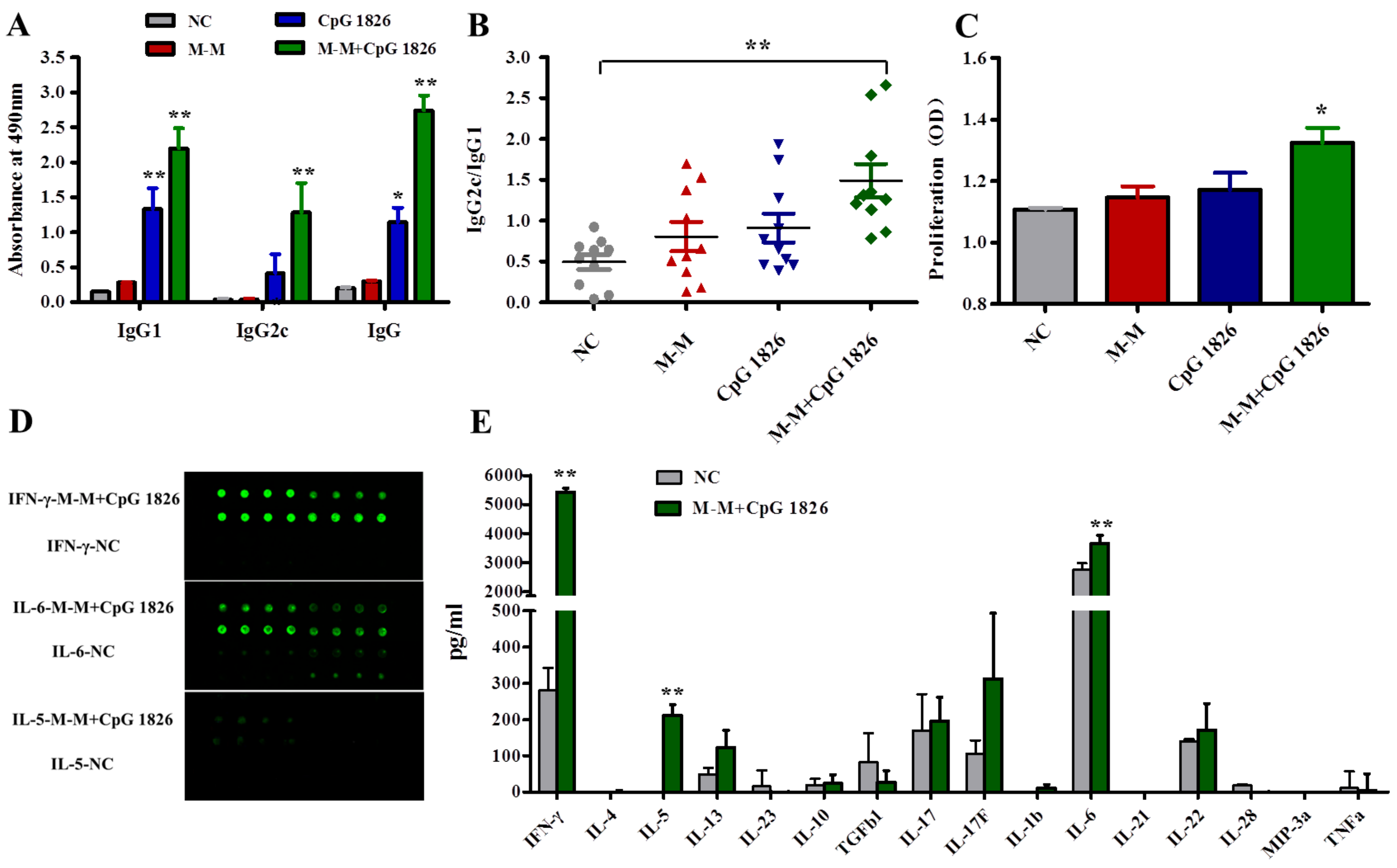
2.2. M-M and CpG 1826 Enhance the Antitumor Response by Inducing MUC1-Specific Humoral and Cellular Immune Responses
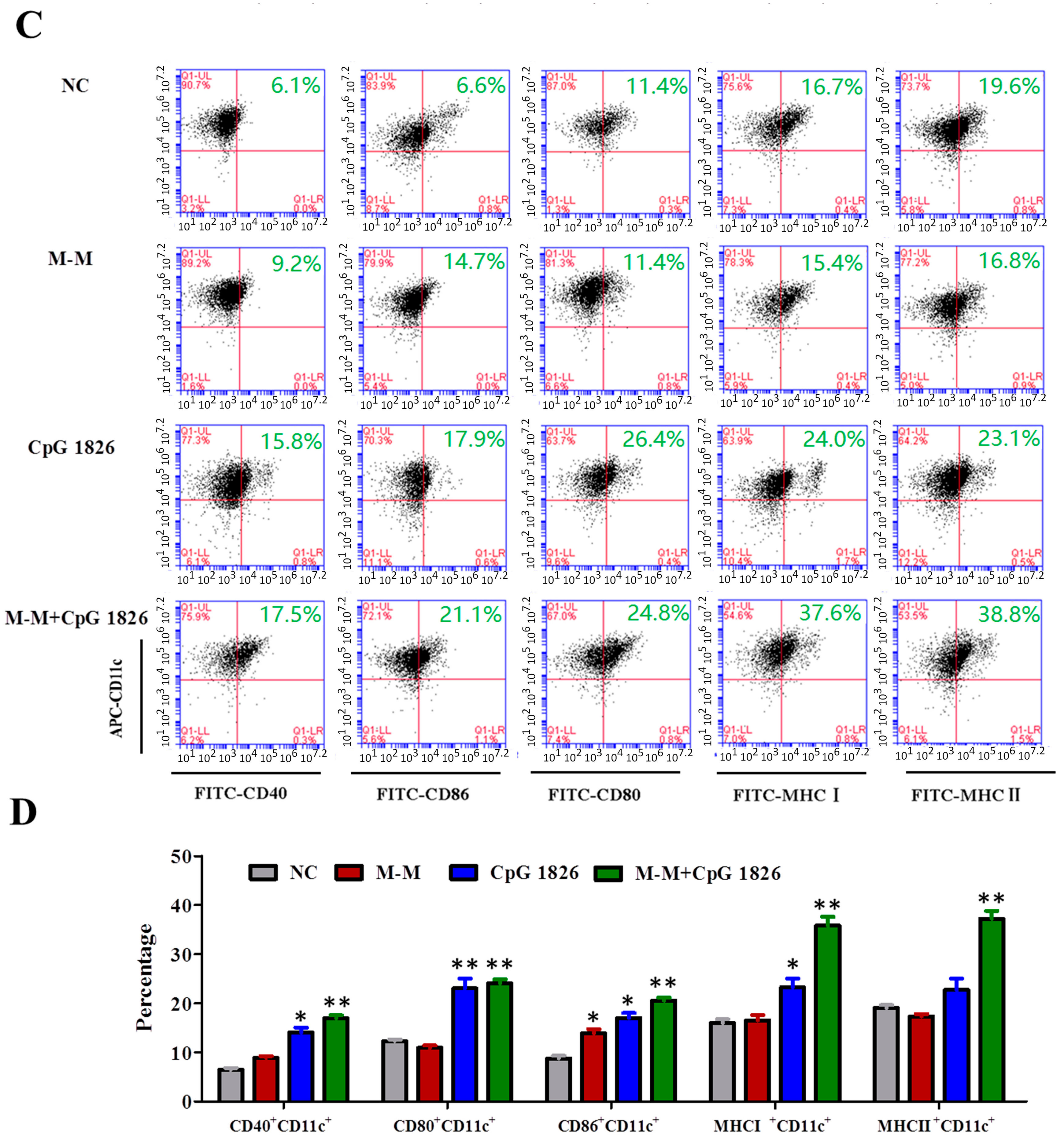
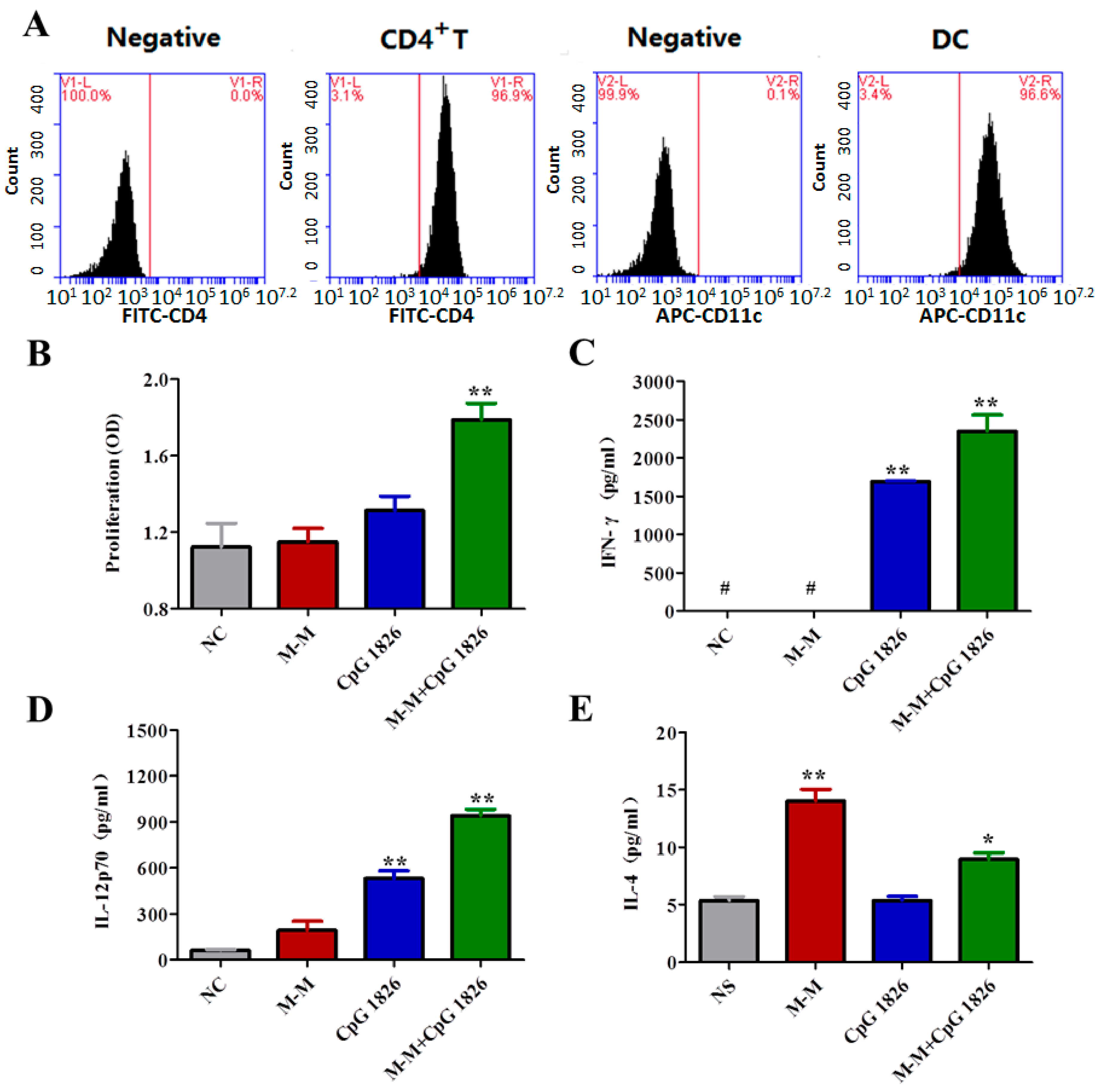
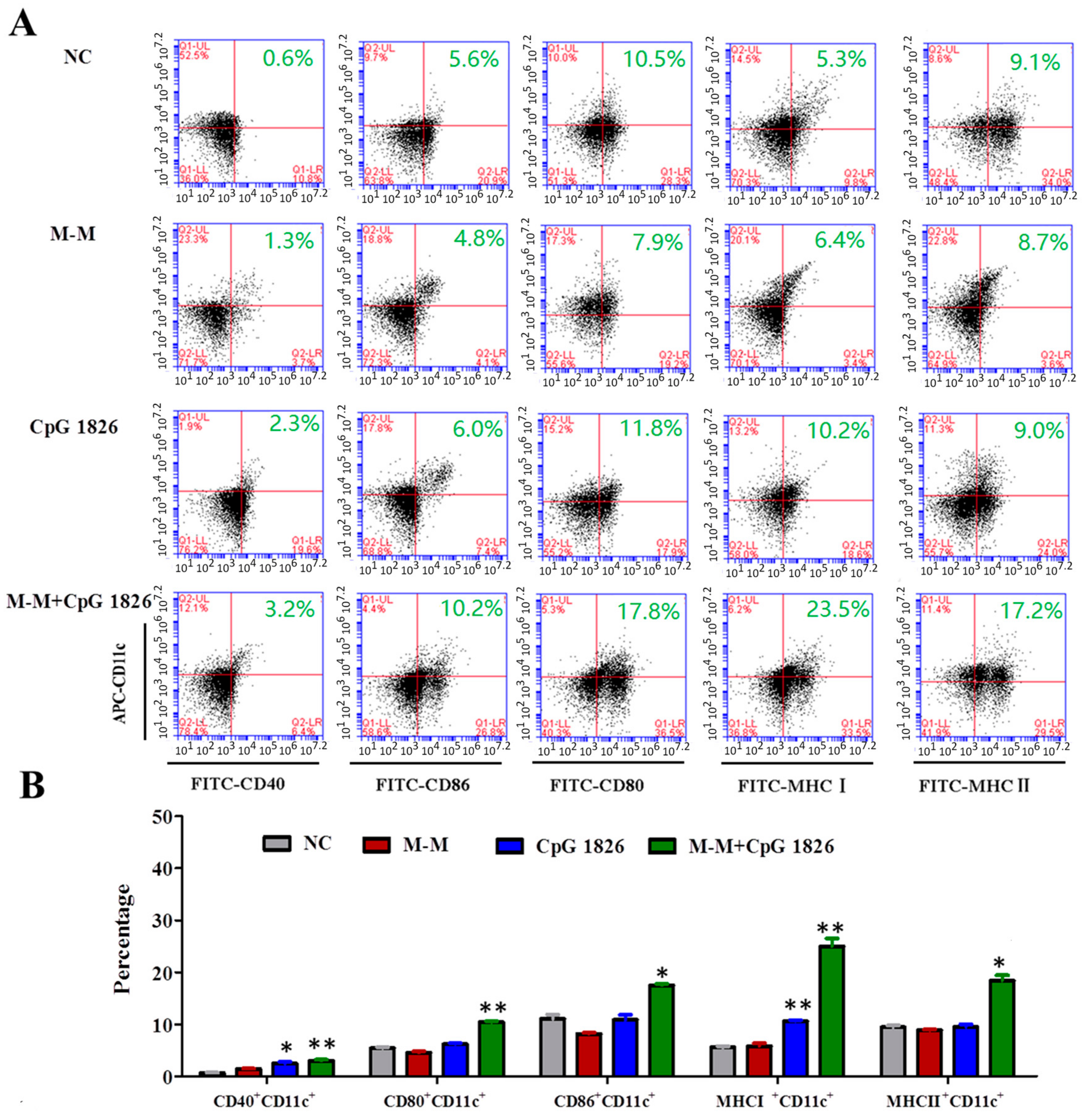
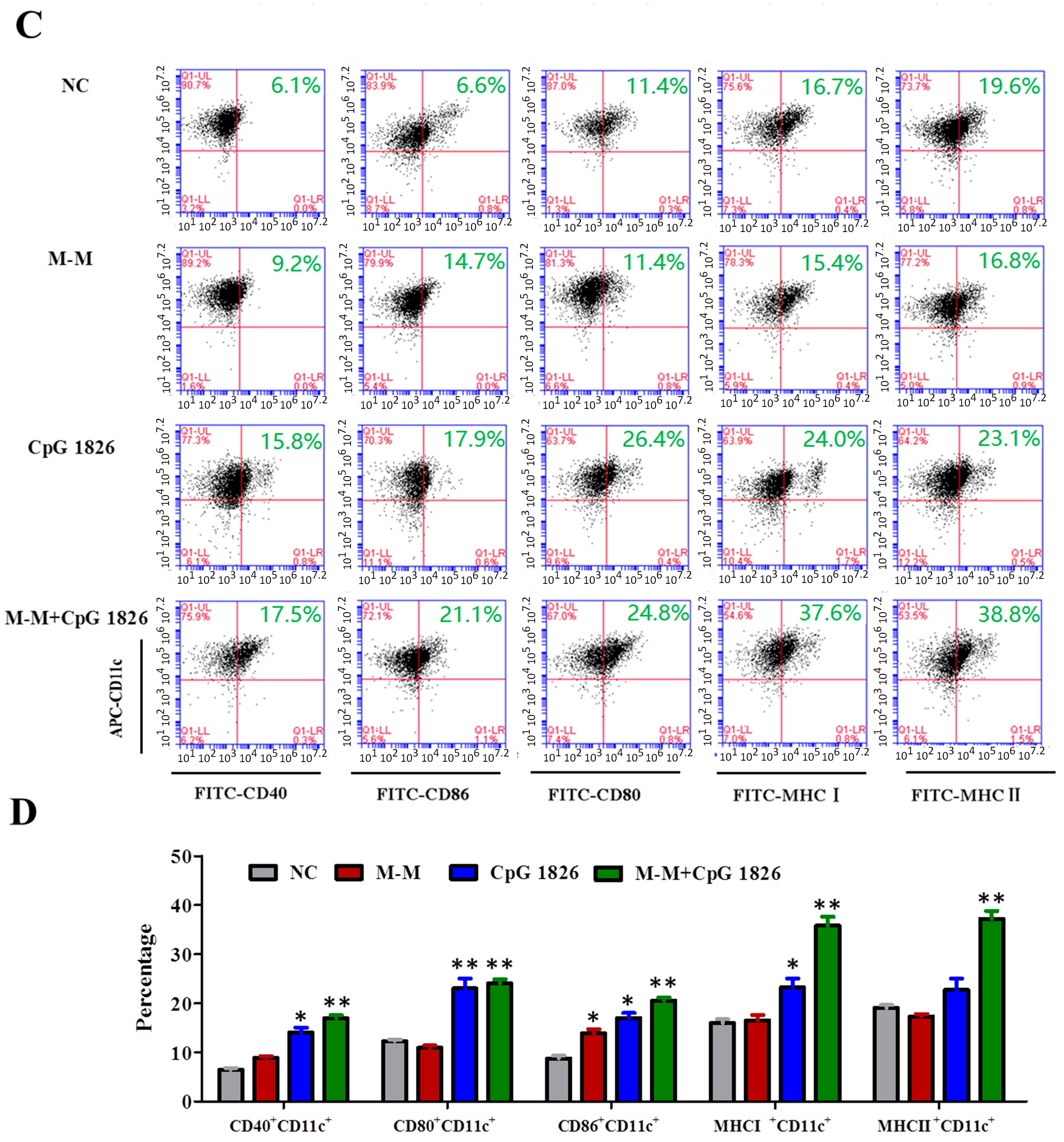
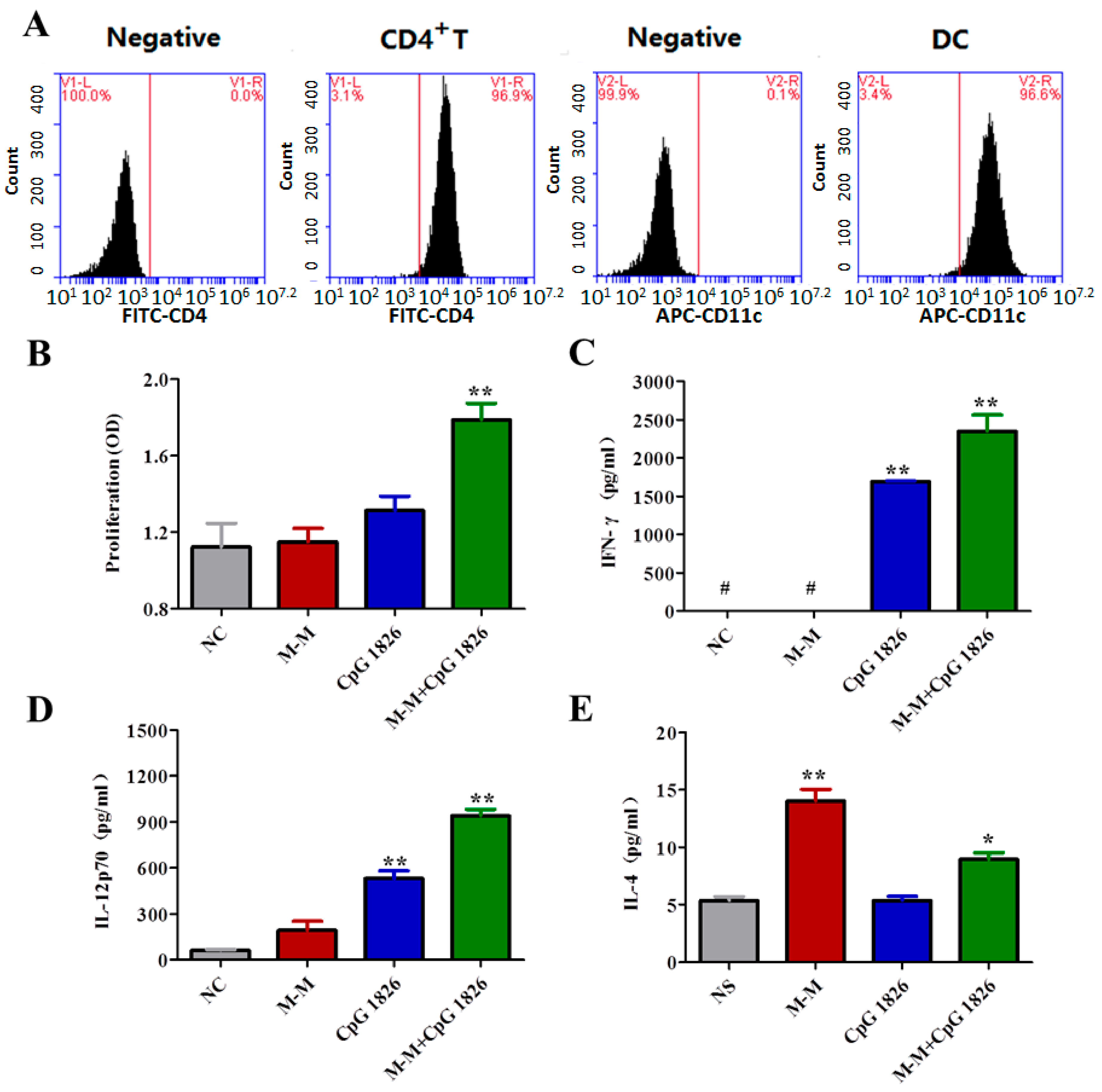
2.3. M-M Combined with CpG 1826 Promote DC Maturation In Vivo and In Vitro
2.4. Combination of M-M and CpG 1826 Promotes the Th1 Polarization of CD4+ T Cell Co-Cultured with BMDCs
2.5. Combination of M-M and CpG 1826 Enhanced the Prophylactic and Therapeutic Antitumor Immune Activity
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Immunization
4.3. Tumor Protection in a Prophylactic Model
4.4. ELISA for MUC1-Specific Immunoglobulin Subclasses
4.5. MUC1 Specific Cell Proliferation and Th Activity Assay
4.6. Cytokine Assay by Quantibody® Array
4.7. MUC1-Specific CTL Cytotoxicity Assay
4.8. Analysis of DC Maturation by Flow Cytometry
4.9. Analysis of the CD4+ T Cell Purity
4.10. Activity Analysis of the CD4+ T Cells Co-Cultured with the BMDCs
4.11. Survival of Mice in the Prophylactic and Therapeutic Model
4.12. Statistical Analysis
Author Contributions
Conflicts of Interest
Abbreviations
| MBP | Maltose-Binding Protein |
| M-M | MUC1-MBP |
| CpG | CpG Oligodeoxynucleotides |
| TLR | Toll-like Receptor |
| B16-MUC1 | MUC1-Overexpressed B16 Melanoma Cells |
| BCG | Bacillus Calmette-Guérin |
| APC | Antigen Presenting Cell |
| BMDCs | Bone Marrow Dendritic Cells |
| mDC | Marrow Derived DCs |
| pDC | Plasmacytoid Dendritic Cells |
| dLNs | Draining Lymph Nodes |
| SI | Stimulation Index |
| CTL | Cytotoxic T Lymphocytes |
References
- Sousa, A.M.; Grandgenett, P.M.; David, L.; Almeida, R.; Hollingsworth, M.A.; Santos-Silva, F. Reflections on MUC1 glycoprotein: The hidden potential of isoforms in carcinogenesis. Acta Pathol. Microbiol. Immunol. Scand. 2016, 124, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Rivalland, G.; Loveland, B.; Mitchell, P. Update on Mucin-1 immunotherapy in cancer: A clinical perspective. Expert Opin. Biol. Ther. 2015, 15, 1773–1787. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.E.; Taylor-Papadimitriou, J.; Burchell, J.M. MUC1 immunotherapy. Immunotherapy 2010, 2, 305–327. [Google Scholar] [CrossRef] [PubMed]
- Guibert, N.; Delaunay, M.; Mazieres, J. Targeting the immune system to treat lung cancer: Rationale and clinical experience. Ther. Adv. Respir. Dis. 2015, 9, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Petrovsky, N.; Aguilar, J.C. Vaccine adjuvants: Current state and future trends. Immunol. Cell Biol. 2004, 82, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.Z.; Duan, G.C.; Fan, Q.T.; Xi, Y.L. Fusion expression of Helicobacter pylori neutrophil-activating protein in E. coli. World J. Gastroenterol. 2005, 11, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.; Palmer, D.R.; Simmons, M.; Sun, P.; Bisbing, J.; McClain, S.; Mani, S.; Burgess, T.; Gunther, V.; Sun, W. Potential role for Toll-like receptor 4 in mediating Escherichia coli maltose-binding protein activation of dendritic cells. Infect. Immun. 2007, 75, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ni, W.; Liu, G.; Wang, J.; Xie, F.; Yuan, H.; Guo, Y.; Zhai, R.; Chen, T.; Li, Q.; et al. Escherichia coli maltose-binding protein (MBP) directly induces mouse Th1 activation through upregulating TLR2 and downregulating TLR4 expressions. Immunobiology 2015, 220, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ma, J.; Fang, F.; Zhou, J.; Song, X.; Liu, Z.; Tai, G. Effect of Escherichia coli maltose-binding protein on mouse Th1 cell activation. Chin. J. Immunol. 2009, 25, 504–507. [Google Scholar]
- Ni, W.; Zhang, Q.; Liu, G.; Wang, F.; Yuan, H.; Guo, Y.; Zhang, X.; Xie, F.; Li, Q.; Tai, G. Escherichia coli maltose-binding protein activates mouse peritoneal macrophages and induces M1 polarization via TLR2/4 in vivo and in vitro. Int. Immunopharmacol. 2014, 21, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Wang, F.; Liu, G.; Zhang, N.; Yuan, H.; Jie, J.; Tai, G. TLR9 played a more important role than TLR2 in the combination of maltose-binding protein and BCG-induced Th1 activation. Mol. Immunol. 2016, 79, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Redelman-Sidi, G.; Glickman, M.S.; Bochner, B.H. The mechanism of action of BCG therapy for bladder cancer—A current perspective. Nat. Rev. Urol. 2014, 11, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Ma, J.; Ni, W.; Wang, F.; Sun, X.; Li, Y.; Li, Q.; Xie, F.; Wang, J.; Zhai, R.; et al. MUC1 and maltose-binding protein recombinant fusion protein combined with Bacillus Calmette-Guerin induces MUC1-specific and nonspecific anti-tumor immunity in mice. Mol. Med. Rep. 2014, 10, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, J.; Guo, Y.; Chen, T.; Ni, W.; Yuan, H.; Zhang, N.; Xie, F.; Tai, G. Pre-clinical toxicity and immunogenicity evaluation of a MUC1-MBP/BCG anti-tumor vaccine. Int. Immunopharmacol. 2016, 33, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Brausi, M.; Oddens, J.; Sylvester, R.; Bono, A.; van de Beek, C.; van Andel, G.; Gontero, P.; Turkeri, L.; Marreaud, S.; Collette, S.; et al. Side effects of Bacillus Calmette-Guerin (BCG) in the treatment of intermediate- and high-risk Ta, T1 papillary carcinoma of the bladder: Results of the EORTC genito-urinary cancers group randomised phase 3 study comparing one-third dose with full dose and 1 year with 3 years of maintenance BCG. Eur. Urol. 2014, 65, 69–76. [Google Scholar] [PubMed]
- Corbel, M.J. Reasons for instability of bacterial vaccines. Dev. Biol. Stand. 1996, 87, 113–124. [Google Scholar] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Heikenwalder, M.; Polymenidou, M.; Junt, T.; Sigurdson, C.; Wagner, H.; Akira, S.; Zinkernagel, R.; Aguzzi, A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat. Med. 2004, 10, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, O.S.; Lohse, N.; Harboe, Z.B.; Offersen, R.; Bukh, A.R.; Davis, H.L.; Schonheyder, H.C.; Ostergaard, L. Improving the immunogenicity of pneumococcal conjugate vaccine in HIV-infected adults with a toll-like receptor 9 agonist adjuvant: A randomized, controlled trial. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 51, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Valmori, D.; Souleimanian, N.E.; Tosello, V.; Bhardwaj, N.; Adams, S.; O’Neill, D.; Pavlick, A.; Escalon, J.B.; Cruz, C.M.; Angiulli, A.; et al. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc. Natl. Acad. Sci. USA 2007, 104, 8947–8952. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Weeratna, R.D.; McCluskie, M.J.; Xu, Y.; Davis, H.L. CpG DNA induces stronger immune responses with less toxicity than other adjuvants. Vaccine 2000, 18, 1755–1762. [Google Scholar] [CrossRef]
- Vollmer, J.; Weeratna, R.; Payette, P.; Jurk, M.; Schetter, C.; Laucht, M.; Wader, T.; Tluk, S.; Liu, M.; Davis, H.L.; et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 2004, 34, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.S.; Greenberg, N.; Billington, M.; Zhang, L.; DeFilippi, C.; May, R.C.; Bajwa, K.K. Phase 1 testing of detoxified LPS/group B meningococcal outer membrane protein vaccine with and without synthetic CPG 7909 adjuvant for the prevention and treatment of sepsis. Vaccine 2015, 33, 6719–6726. [Google Scholar] [CrossRef] [PubMed]
- Minang, J.T.; Inglefield, J.R.; Harris, A.M.; Lathey, J.L.; Alleva, D.G.; Sweeney, D.L.; Hopkins, R.J.; Lacy, M.J.; Bernton, E.W. Enhanced early innate and T cell-mediated responses in subjects immunized with Anthrax Vaccine Adsorbed Plus CPG 7909 (AV7909). Vaccine 2014, 32, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.S.; Cohen, P.A.; Finn, O.J. Lack of effective MUC1 tumor antigen-specific immunity in MUC1-transgenic mice results from a Th/T regulatory cell imbalance that can be corrected by adoptive transfer of wild-type Th cells. J. Immunol. 2007, 178, 2787–2793. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Williams, M.A.; Bevan, M.J. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 2004, 5, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, L.; Wang, S.; Yang, J.; Yang, X. Enhanced inducible costimulator ligand (ICOS-L) expression on dendritic cells in interleukin-10 deficiency and its impact on T-cell subsets in respiratory tract infection. Mol. Med. 2013, 19, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Battiany, J.; Poeck, H.; Wagner, M.; Kerkmann, M.; Lubenow, N.; Rothenfusser, S.; Endres, S. Rational design of new CpG oligonucleotides that combine B cell activation with high IFN-α induction in plasmacytoid dendritic cells. Eur. J. Immunol. 2003, 33, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Salio, M.; Cella, M.; Vermi, W.; Facchetti, F.; Palmowski, M.J.; Smith, C.L.; Shepherd, D.; Colonna, M.; Cerundolo, V. Plasmacytoid dendritic cells prime IFN-gamma-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. Eur. J. Immunol. 2003, 33, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Molenkamp, B.G.; van Leeuwen, P.A.; Meijer, S.; Sluijter, B.J.; Wijnands, P.G.; Baars, A.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Intradermal CpG-B activates both plasmacytoid and myeloid dendritic cells in the sentinel lymph node of melanoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Sparwasser, T.; Koch, E.S.; Vabulas, R.M.; Heeg, K.; Lipford, G.B.; Ellwart, J.W.; Wagner, H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 1998, 28, 2045–2054. [Google Scholar] [CrossRef]
- Ding, C.; Wang, L.; Marroquin, J.; Yan, J. Targeting of antigens to B cells augments antigen-specific T-cell responses and breaks immune tolerance to tumor-associated antigen MUC1. Blood 2008, 112, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Gatard-Scheikl, T.; Kintz, J.; Gantzer, M.; Schaedler, E.; Rittner, K.; Cochin, S.; Fournel, S.; Preville, X. Intravenous injection of MVA virus targets CD8+ lymphocytes to tumors to control tumor growth upon combinatorial treatment with a TLR9 agonist. Cancer Immunol. Res. 2014, 2, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M.; Klaschik, S.; Sato, T.; Tross, D. CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases. Adv. Drug Deliv. Rev. 2009, 61, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Kuzel, T.; Drucker, B.J.; Urba, W.J.; Bukowski, R.M. Safety and efficacy of PF-3512676 for the treatment of stage IV renal cell carcinoma: An open-label, multicenter phase I/II study. Clin. Genitourin. Cancer 2009, 7, E58–E65. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.; Rossner, S.; Koch, F.; Romani, N.; Schuler, G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jie, J.; Zhang, Y.; Zhou, H.; Zhai, X.; Zhang, N.; Yuan, H.; Ni, W.; Tai, G. CpG ODN1826 as a Promising Mucin1-Maltose-Binding Protein Vaccine Adjuvant Induced DC Maturation and Enhanced Antitumor Immunity. Int. J. Mol. Sci. 2018, 19, 920. https://doi.org/10.3390/ijms19030920
Jie J, Zhang Y, Zhou H, Zhai X, Zhang N, Yuan H, Ni W, Tai G. CpG ODN1826 as a Promising Mucin1-Maltose-Binding Protein Vaccine Adjuvant Induced DC Maturation and Enhanced Antitumor Immunity. International Journal of Molecular Sciences. 2018; 19(3):920. https://doi.org/10.3390/ijms19030920
Chicago/Turabian StyleJie, Jing, Yixin Zhang, Hongyue Zhou, Xiaoyu Zhai, Nannan Zhang, Hongyan Yuan, Weihua Ni, and Guixiang Tai. 2018. "CpG ODN1826 as a Promising Mucin1-Maltose-Binding Protein Vaccine Adjuvant Induced DC Maturation and Enhanced Antitumor Immunity" International Journal of Molecular Sciences 19, no. 3: 920. https://doi.org/10.3390/ijms19030920
APA StyleJie, J., Zhang, Y., Zhou, H., Zhai, X., Zhang, N., Yuan, H., Ni, W., & Tai, G. (2018). CpG ODN1826 as a Promising Mucin1-Maltose-Binding Protein Vaccine Adjuvant Induced DC Maturation and Enhanced Antitumor Immunity. International Journal of Molecular Sciences, 19(3), 920. https://doi.org/10.3390/ijms19030920