Agnoprotein Is an Essential Egress Factor during BK Polyomavirus Infection

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
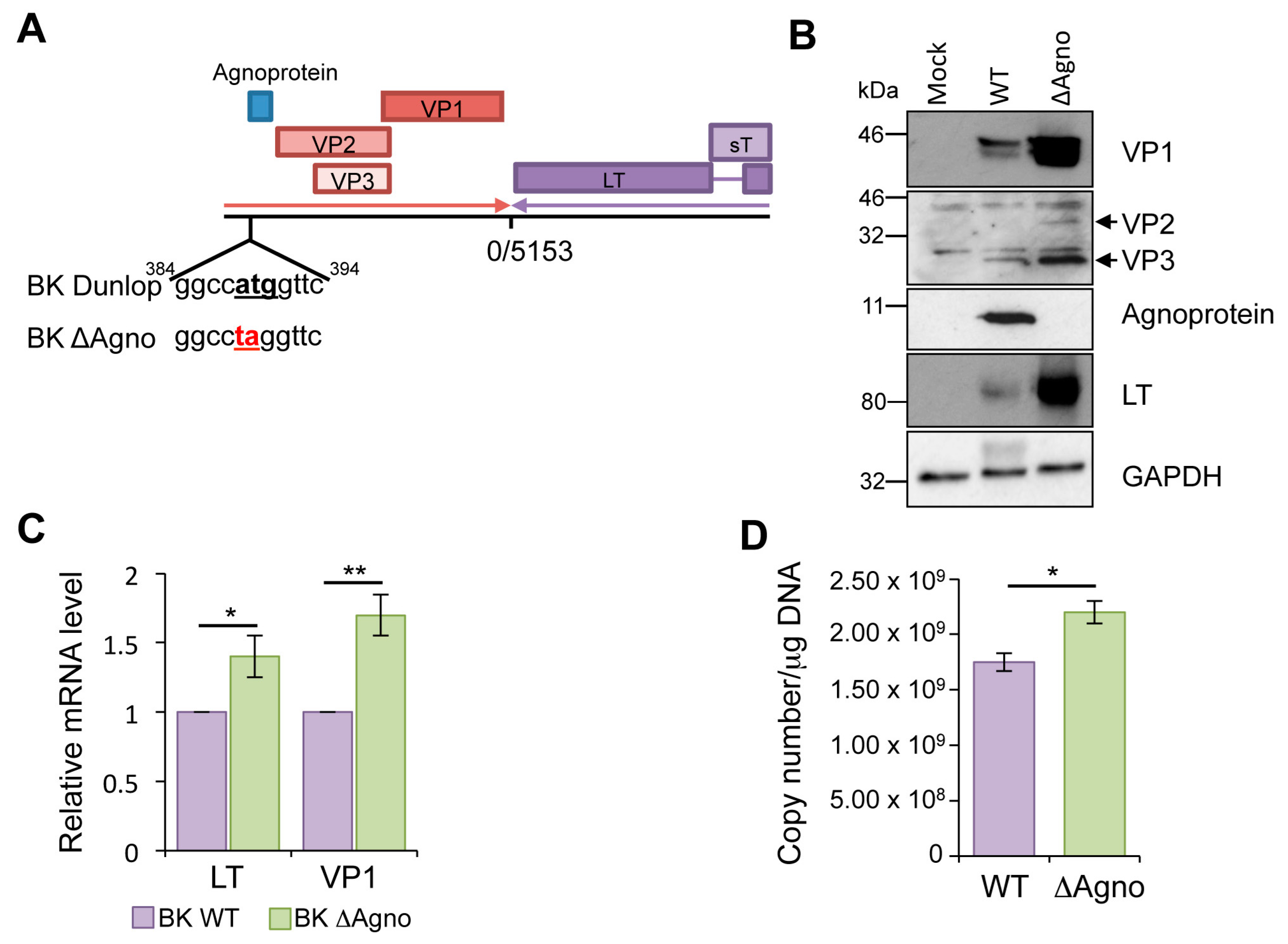
2.1. Loss of Agnoprotein Increases BK Transcription and Protein Expression
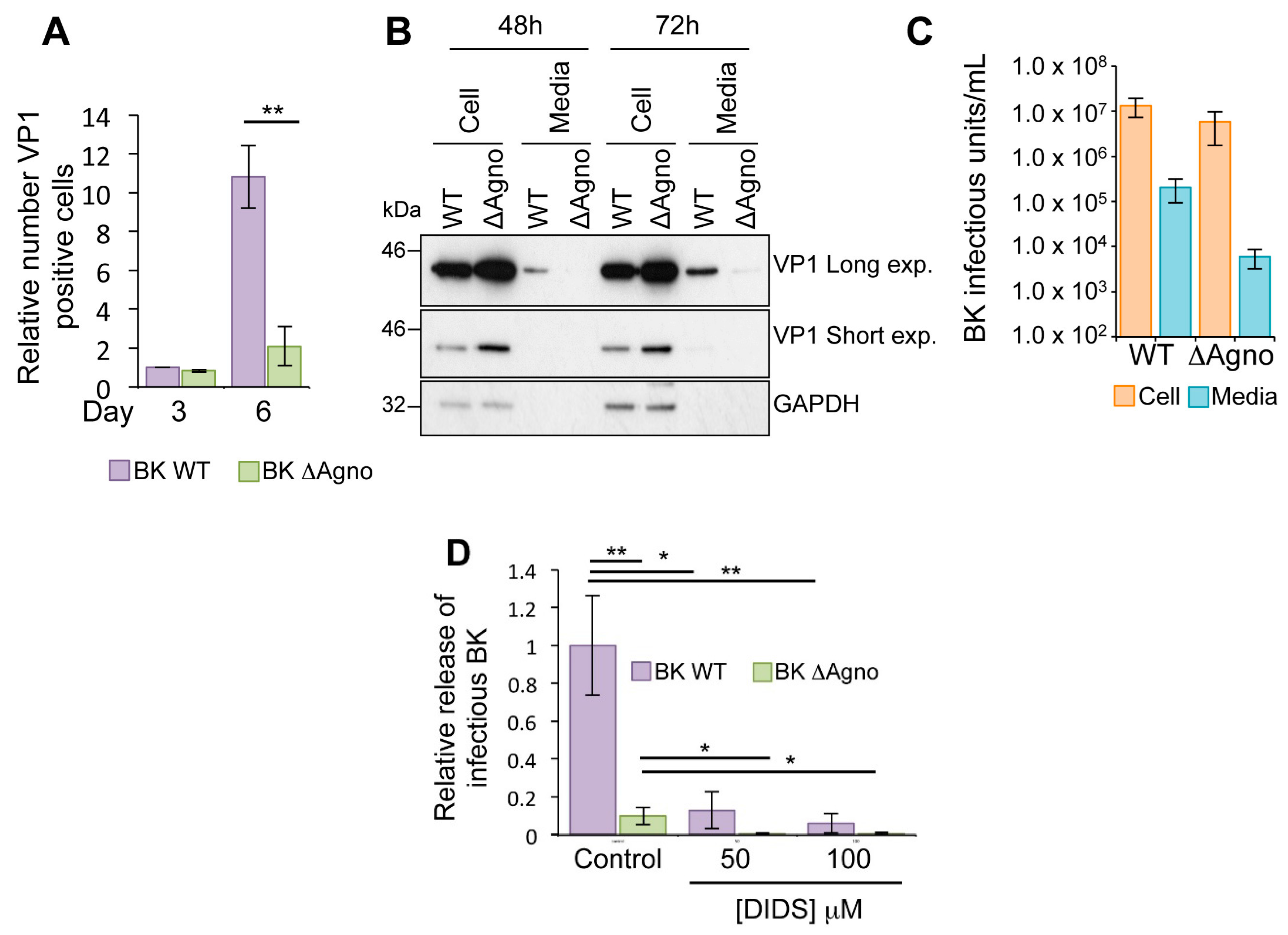
2.2. Agnoprotein Is Required for BK Virus Release
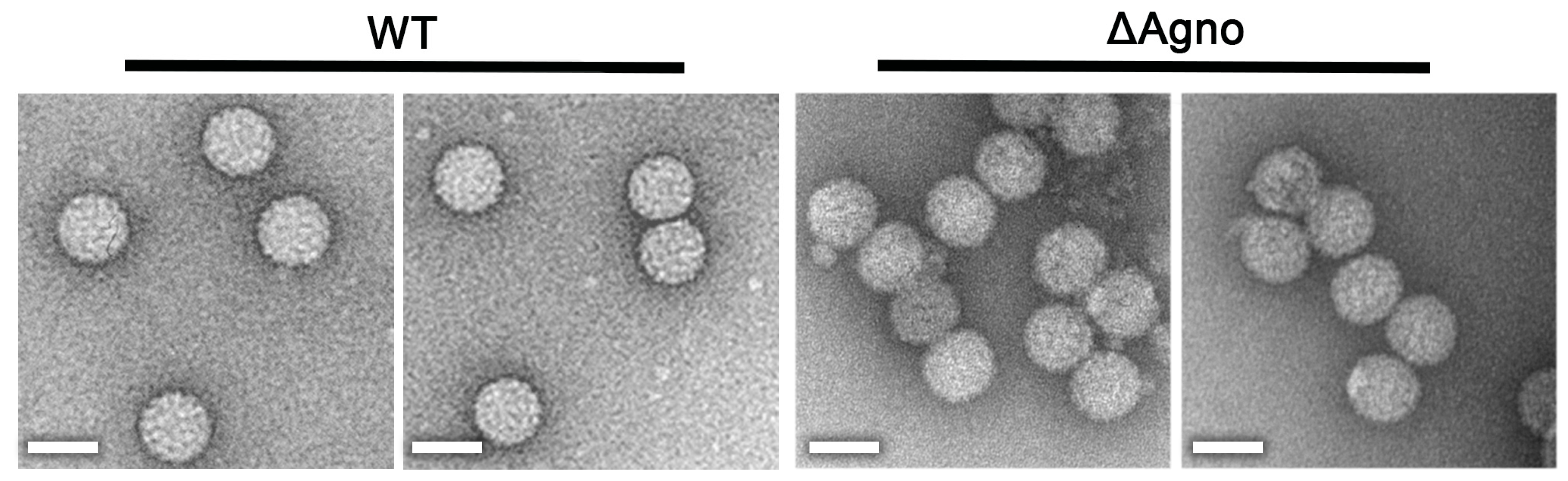
2.3. Agnoprotein Is Not Required for the Production of BK Virions
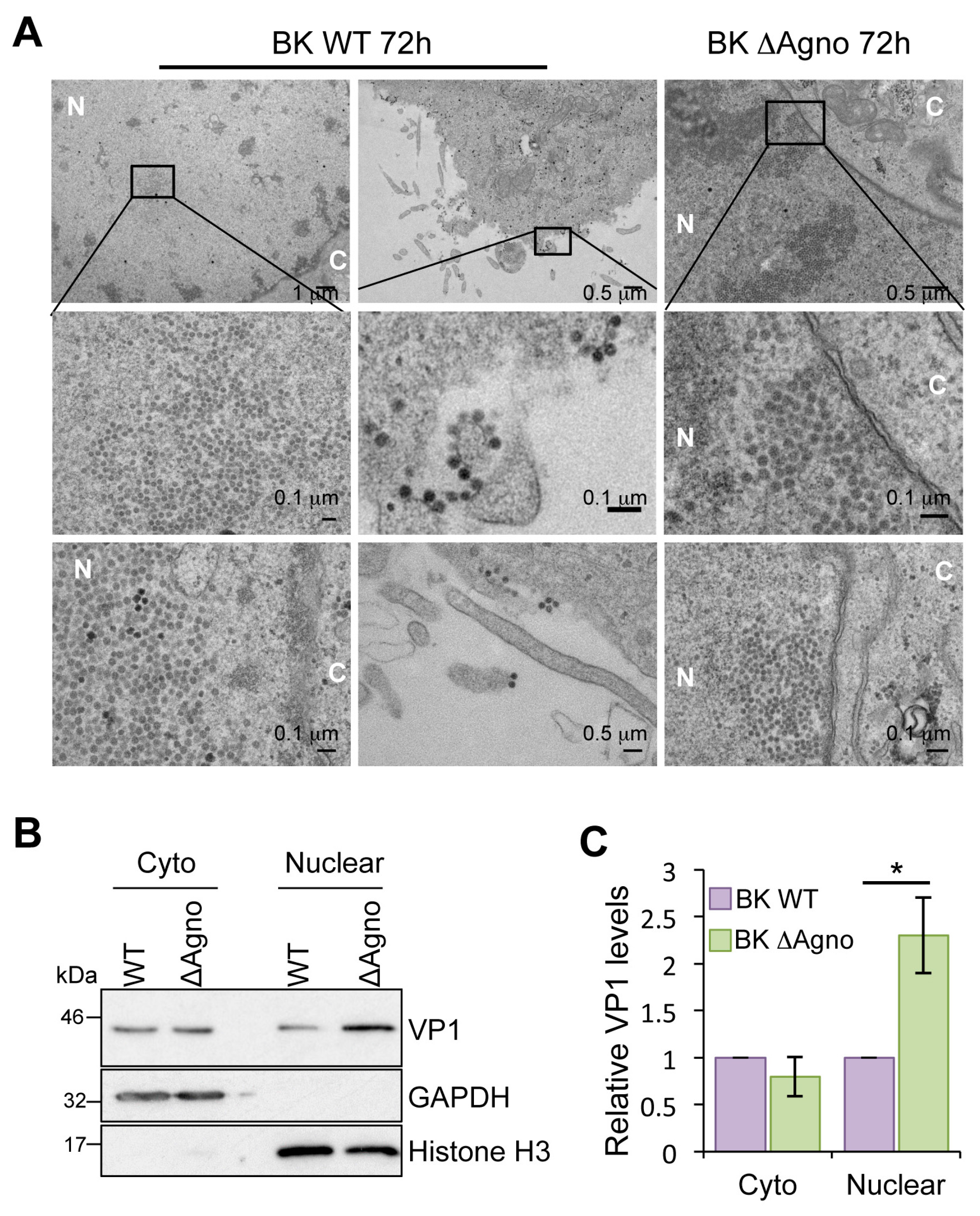
2.4. Agnoprotein Is Required for the Nuclear Egress of BK Particles
2.5. Agnoprotein Does Not Cause Gross Destabilization of the Nuclear Membrane
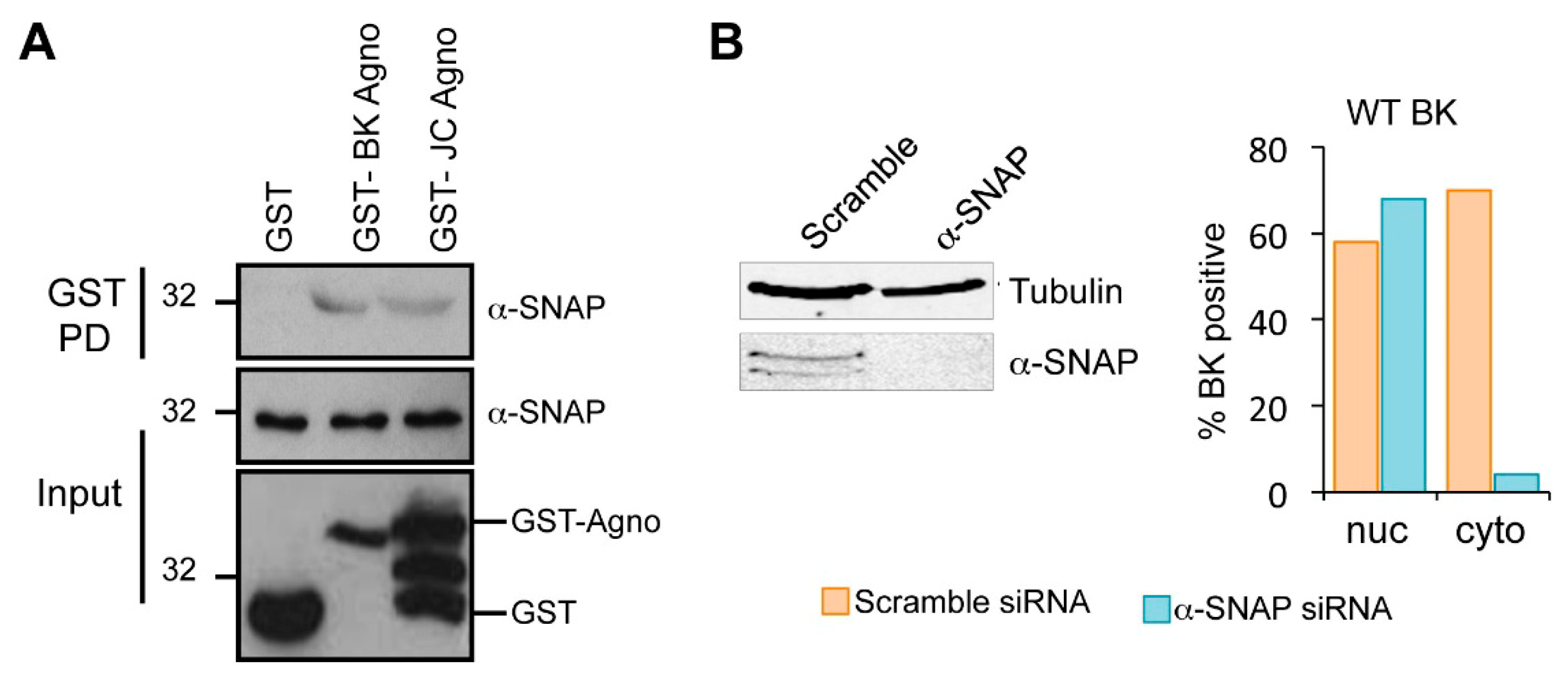
2.6. Host α-SNAP Is Necessary for BK Egress
3. Discussion
4. Methods and Materials
4.1. Cell Culture
4.2. Generation of an Agnoprotein Knockout BK Dunlop Genome
4.3. Transfection of Virus Genomes
4.4. Virus Culture and Purification
4.5. Cell Infections and Harvesting Virus
4.6. Fluorescent Focus Unit Assay Using IncuCyte ZOOM Analysis
4.7. Immunofluorescence
4.8. Western Blotting
4.9. Quantitative PCR
4.10. Quantitative Reverse Transcriptase PCR
4.11. Electron Microscopy
4.12. Transmission Electron Microscopy in Cells
4.13. Cell Fractionation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buck, C.B.; van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The Ancient Evolutionary History of Polyomaviruses. PLoS Pathog. 2016, 12, e1005574. [Google Scholar] [CrossRef] [PubMed]
- Peretti, A.; FitzGerald, P.C.; Bliskovsky, V.; Pastrana, D.V.; Buck, C.B. Genome Sequence of a Fish-Associated Polyomavirus, Black Sea Bass (Centropristis striata) Polyomavirus 1. Genome Announc. 2015, 3, e01476-14. [Google Scholar] [CrossRef] [PubMed]
- Peretti, A.; FitzGerald, P.C.; Bliskovsky, V.; Buck, C.B.; Pastrana, D.V. Hamburger polyomaviruses. J. Gen. Virol. 2015, 96, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Hurdiss, D.L.; Morgan, E.L.; Thompson, R.F.; Prescott, E.L.; Panou, M.M.; Macdonald, A.; Ranson, N.A. New Structural Insights into the Genome and Minor Capsid Proteins of BK Polyomavirus using Cryo-Electron Microscopy. Structure 2016, 24, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Publ. Group 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, E.; Janssens, R.W.A.; Lauber, C.; Bouwes Bavinck, J.N.; Gorbalenya, A.E.; Feltkamp, M.C.W. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010, 6, e1001024. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 1, 1253–1257. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Knowles, W.A. Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV). Adv. Exp. Med. Biol. 2006, 577, 19–45. [Google Scholar] [PubMed]
- Bennett, S.M.; Broekema, N.M.; Imperiale, M.J. BK polyomavirus: Emerging pathogen. Microbes Infect. 2012, 14, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Köhli, S.; Dickenmann, M.; Hirsch, H.H. Inhibition of polyomavirus BK-specific T-Cell responses by immunosuppressive drugs. Transplantation 2009, 88, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Shah, K.V. Polyomaviruses and human diseases. Adv. Exp. Med. Biol. 2006, 577, 1–18. [Google Scholar] [PubMed]
- Dropulic, L.K.; Jones, R.J. Polyomavirus BK infection in blood and marrow transplant recipients. Bone Marrow Transpl. 2008, 41, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Balba, G.P.; Javaid, B.; Timpone, J.G. BK polyomavirus infection in the renal transplant recipient. Infect. Dis. Clin. N. Am. 2013, 27, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Drachenberg, C.B.; Wali, R.; Hirsch, H.H. The decade of polyomavirus BK-associated nephropathy: State of affairs. Transplantation 2009, 87, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Safrin, S.; Cherrington, J.; Jaffe, H. Clinical uses of cidofovir. Rev. Med. Virol. 1997, 7, 145–156. [Google Scholar] [CrossRef]
- Kuypers, D.R.J. Management of polyomavirus-associated nephropathy in renal transplant recipients. Nat. Rev. Nephrol. 2012, 8, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.M.; Gupta, G.; Vats, A.; Shapiro, R.; Randhawa, P.S. Polyomavirus BK non-coding control region rearrangements in health and disease. J. Med. Virol. 2007, 79, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B. Exposing the Molecular Machinery of BK Polyomavirus. Structure 2016, 24, 495. [Google Scholar] [CrossRef] [PubMed]
- Gerits, N.; Moens, U. Agnoprotein of mammalian polyomaviruses. Virology 2012, 432, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, C.H.; Traavik, T.; Hey, A. The agnogene of the human polyomavirus BK is expressed. J. Virol. 1998, 72, 6233–6236. [Google Scholar] [PubMed]
- Unterstab, G.; Gosert, R.; Leuenberger, D.; Lorentz, P.; Rinaldo, C.H.; Hirsch, H.H. The polyomavirus BK agnoprotein co-localizes with lipid droplets. Virology 2010, 399, 322–331. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Johannessen, M.; Myhre, M.R.; Dragset, M.; Tümmler, C.; Moens, U. Phosphorylation of human polyomavirus BK agnoprotein at Ser-11 is mediated by PKC and has an important regulative function. Virology 2008, 379, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sariyer, I.K.; Khalili, K.; Safak, M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: Inhibition by small t antigen. Virology 2008, 375, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Khalili, K.; White, M.K.; Sawa, H.; Nagashima, K.; Safak, M. The agnoprotein of polyomaviruses: A multifunctional auxiliary protein. J. Cell Physiol. 2005, 204, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Orba, Y.; Okada, Y.; Sunden, Y.; Kimura, T.; Tanaka, S.; Nagashima, K.; Hall, W.W.; Sawa, H. The Human Polyoma JC Virus Agnoprotein Acts as a Viroporin. PLoS Pathog. 2010, 6, e1000801. [Google Scholar] [CrossRef] [PubMed]
- Akan, I.; Sariyer, I.K.; Biffi, R.; Palermo, V.; Woolridge, S.; White, M.K.; Amini, S.; Khalili, K.; Safak, M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology 2006, 349, 66–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barkan, A.; Welch, R.C.; Mertz, J.E. Missense mutations in the VP1 gene of simian virus 40 that compensate for defects caused by deletions in the viral agnogene. J. Virol. 1987, 61, 3190–3198. [Google Scholar] [PubMed]
- Sedman, S.A.; Good, P.J.; Mertz, J.E. Leader-encoded open reading frames modulate both the absolute and relative rates of synthesis of the virion proteins of simian virus 40. J. Virol. 1989, 63, 3884–3893. [Google Scholar] [PubMed]
- Safak, M.; Khalili, K. Physical and functional interaction between viral and cellular proteins modulate JCV gene transcription. J. Neurovirol. 2001, 7, 288–292. [Google Scholar] [PubMed]
- Carswell, S.; Alwine, J.C. Simian virus 40 agnoprotein facilitates perinuclear-nuclear localization of VP1, the major capsid protein. J. Virol. 1986, 60, 1055–1061. [Google Scholar] [PubMed]
- Carswell, S.; Resnick, J.; Alwine, J.C. Construction and characterization of CV-1P cell lines which constitutively express the simian virus 40 agnoprotein: Alteration of plaquing phenotype of viral agnogene mutants. J. Virol. 1986, 60, 415–422. [Google Scholar] [PubMed]
- Sariyer, I.K.; Saribas, A.S.; White, M.K.; Safak, M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol. J. 2011, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Myhre, M.R.; Olsen, G.-H.; Gosert, R.; Hirsch, H.H.; Rinaldo, C.H. Clinical polyomavirus BK variants with agnogene deletion are non-functional but rescued by trans-complementation. Virology 2010, 398, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Stewart, H.; Bartlett, C.; Ross-Thriepland, D.; Shaw, J.; Griffin, S.; Harris, M. A novel method for the measurement of hepatitis C virus infectious titres using the IncuCyte ZOOM and its application to antiviral screening. J. Virol. Methods 2015, 218, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.L.; Caller, L.G.; Foster, V.; Crump, C.M. Anion homeostasis is important for non-lytic release of BK polyomavirus from infected cells. Open Biol. 2015, 5, 150041. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wang, K.; Yu, W.; Lu, W.; Xu, K.; Wang, J.; Ye, B.; Schwarz, W.; Jin, Q.; Sun, B. DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res. 2011, 21, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Royle, J.; Dobson, S.J.; Müller, M.; Macdonald, A. Emerging Roles of Viroporins Encoded by DNA Viruses: Novel Targets for Antivirals? Viruses 2015, 7, 5375–5387. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.; Griffin, S.D.C. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Semba, S.; Sunden, Y.; Orba, Y.; Kobayashi, S.; Nagashima, K.; Kimura, T.; Hasegawa, H.; Sawa, H. Role of JC virus agnoprotein in virion formation. Microbiol. Immunol. 2012, 56, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.S.; Enderlein, D.; Nelson, C.D.S.; Carter, W.S.; Kawano, M.; Xing, L.; Swenson, R.D.; Olson, N.H.; Baker, T.S.; Cheng, R.H.; et al. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology 2011, 411, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Suzuki, T.; Sunden, Y.; Orba, Y.; Kose, S.; Imamoto, N.; Takahashi, H.; Tanaka, S.; Hall, W.W.; Nagashima, K.; et al. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: Role in nuclear egress of viral particles. EMBO Rep. 2005, 6, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Resnick, J.; Shenk, T. Simian virus 40 agnoprotein facilitates normal nuclear location of the major capsid polypeptide and cell-to-cell spread of virus. J. Virol. 1986, 60, 1098–1106. [Google Scholar] [PubMed]
- Johannessen, M.; Walquist, M.; Gerits, N.; Dragset, M.; Spang, A.; Moens, U. BKV agnoprotein interacts with α-soluble N-ethylmaleimide-sensitive fusion attachment protein, and negatively influences transport of VSVG-EGFP. PLoS ONE 2011, 6, e24489. [Google Scholar] [CrossRef] [PubMed]
- Clayson, E.T.; Brando, L.V.; Compans, R.W. Release of simian virus 40 virions from epithelial cells is polarized and occurs without cell lysis. J. Virol. 1989, 63, 2278–2288. [Google Scholar] [PubMed]
- Gerits, N.; Johannessen, M.; Tümmler, C.; Walquist, M.; Kostenko, S.; Snapkov, I.; van Loon, B.; Ferrari, E.; Hübscher, U.; Moens, U. Agnoprotein of polyomavirus BK interacts with proliferating cell nuclear antigen and inhibits DNA replication. Virol. J. 2015, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Orba, Y.; Makino, Y.; Okada, Y.; Sunden, Y.; Hasegawa, H.; Hall, W.W.; Sawa, H. Viroporin activity of the JC polyomavirus is regulated by interactions with the adaptor protein complex 3. Proc. Natl. Acad. Sci. USA 2013, 110, 18668–18673. [Google Scholar] [CrossRef] [PubMed]
- Peter, F.; Wong, S.H.; Subramaniam, V.N.; Tang, B.L.; Hong, W. α-SNAP but not gamma-SNAP is required for ER-Golgi transport after vesicle budding and the Rab1-requiring step but before the EGTA-sensitive step. J. Cell Sci. 1998, 111 Pt 17, 2625–2633. [Google Scholar] [PubMed]
- Barnard, R.J.; Morgan, A.; Burgoyne, R.D. Stimulation of NSF ATPase activity by α-SNAP is required for SNARE complex disassembly and exocytosis. J. Cell Biol. 1997, 139, 875–883. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panou, M.-M.; Prescott, E.L.; Hurdiss, D.L.; Swinscoe, G.; Hollinshead, M.; Caller, L.G.; Morgan, E.L.; Carlisle, L.; Müller, M.; Antoni, M.; et al. Agnoprotein Is an Essential Egress Factor during BK Polyomavirus Infection. Int. J. Mol. Sci. 2018, 19, 902. https://doi.org/10.3390/ijms19030902
Panou M-M, Prescott EL, Hurdiss DL, Swinscoe G, Hollinshead M, Caller LG, Morgan EL, Carlisle L, Müller M, Antoni M, et al. Agnoprotein Is an Essential Egress Factor during BK Polyomavirus Infection. International Journal of Molecular Sciences. 2018; 19(3):902. https://doi.org/10.3390/ijms19030902
Chicago/Turabian StylePanou, Margarita-Maria, Emma L. Prescott, Daniel L. Hurdiss, Gemma Swinscoe, Michael Hollinshead, Laura G. Caller, Ethan L. Morgan, Louisa Carlisle, Marietta Müller, Michelle Antoni, and et al. 2018. "Agnoprotein Is an Essential Egress Factor during BK Polyomavirus Infection" International Journal of Molecular Sciences 19, no. 3: 902. https://doi.org/10.3390/ijms19030902
APA StylePanou, M.-M., Prescott, E. L., Hurdiss, D. L., Swinscoe, G., Hollinshead, M., Caller, L. G., Morgan, E. L., Carlisle, L., Müller, M., Antoni, M., Kealy, D., Ranson, N. A., Crump, C. M., & Macdonald, A. (2018). Agnoprotein Is an Essential Egress Factor during BK Polyomavirus Infection. International Journal of Molecular Sciences, 19(3), 902. https://doi.org/10.3390/ijms19030902