An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
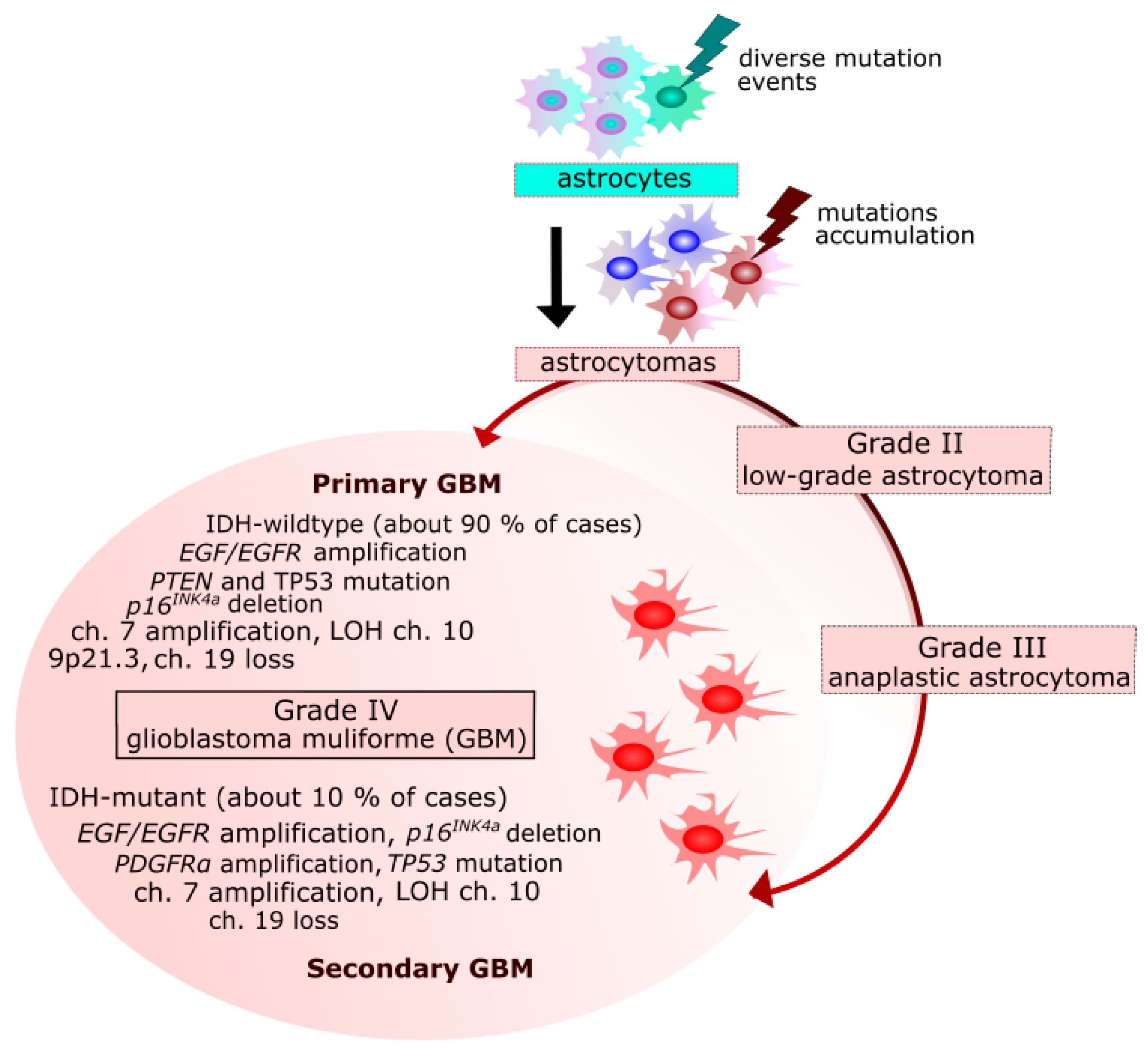
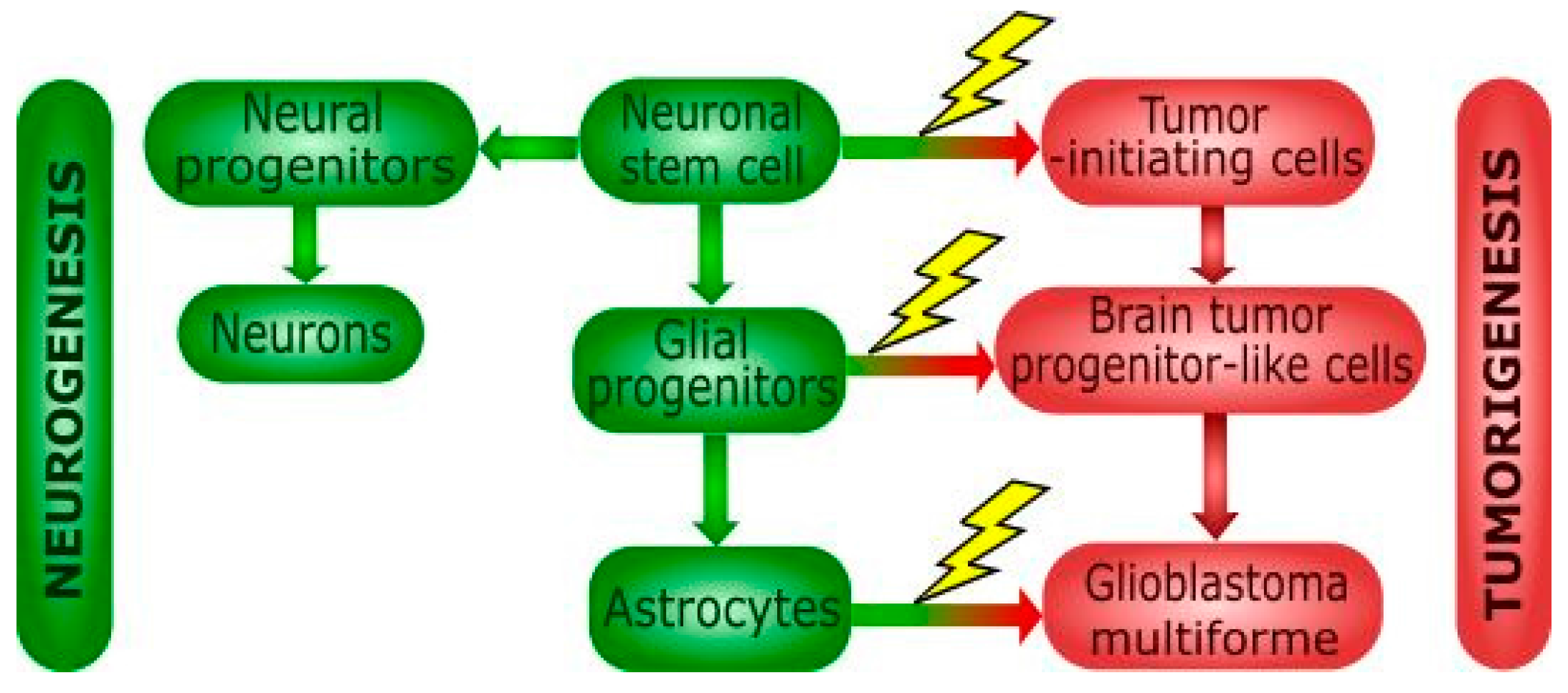
2. Glioblastoma Multiforme
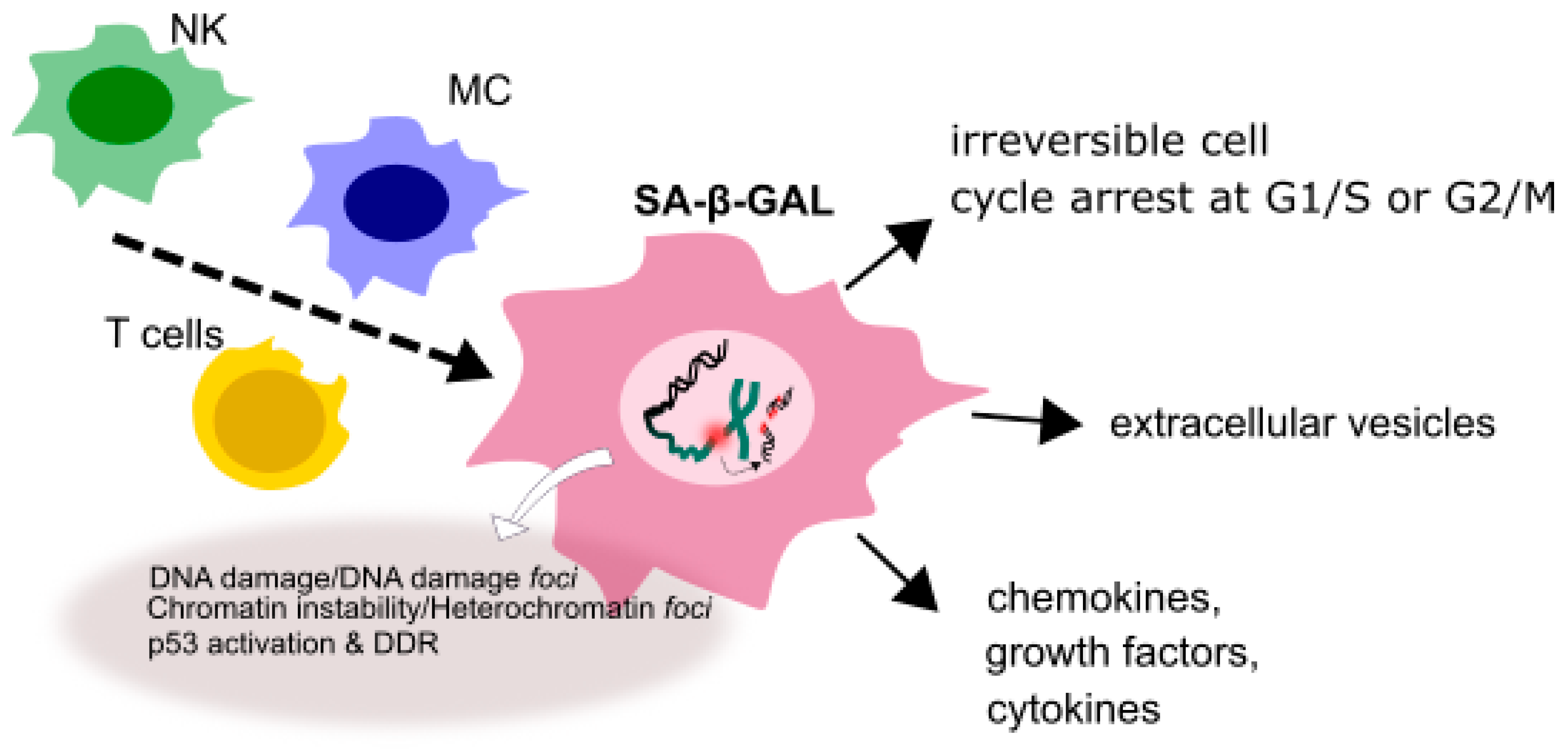
3. Senescence in Glioblastoma
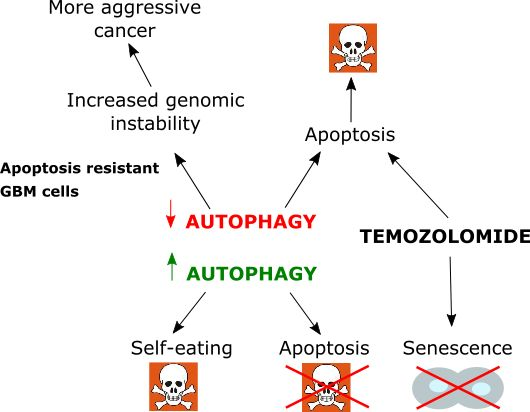
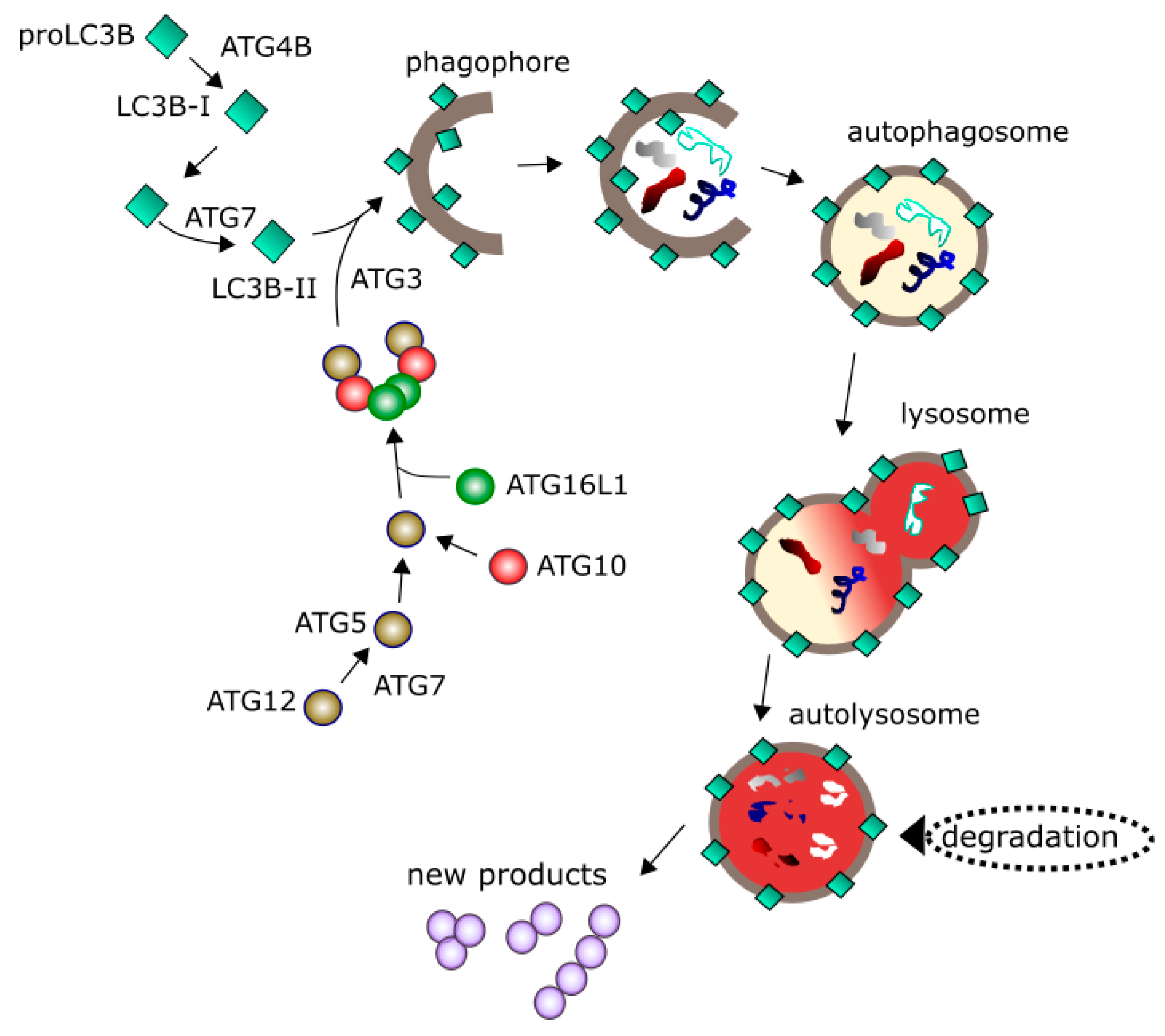
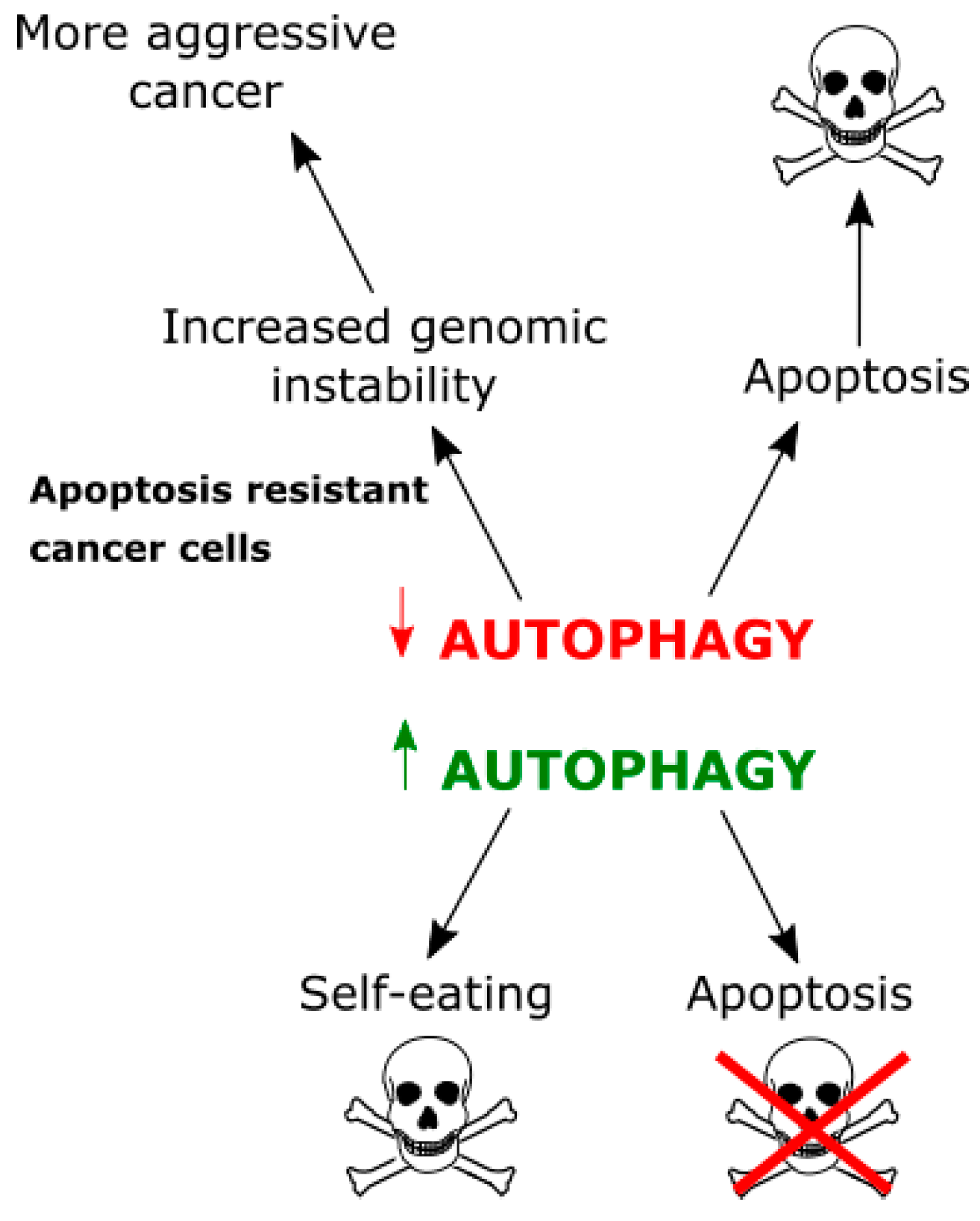
4. Autophagy in Glioblastoma
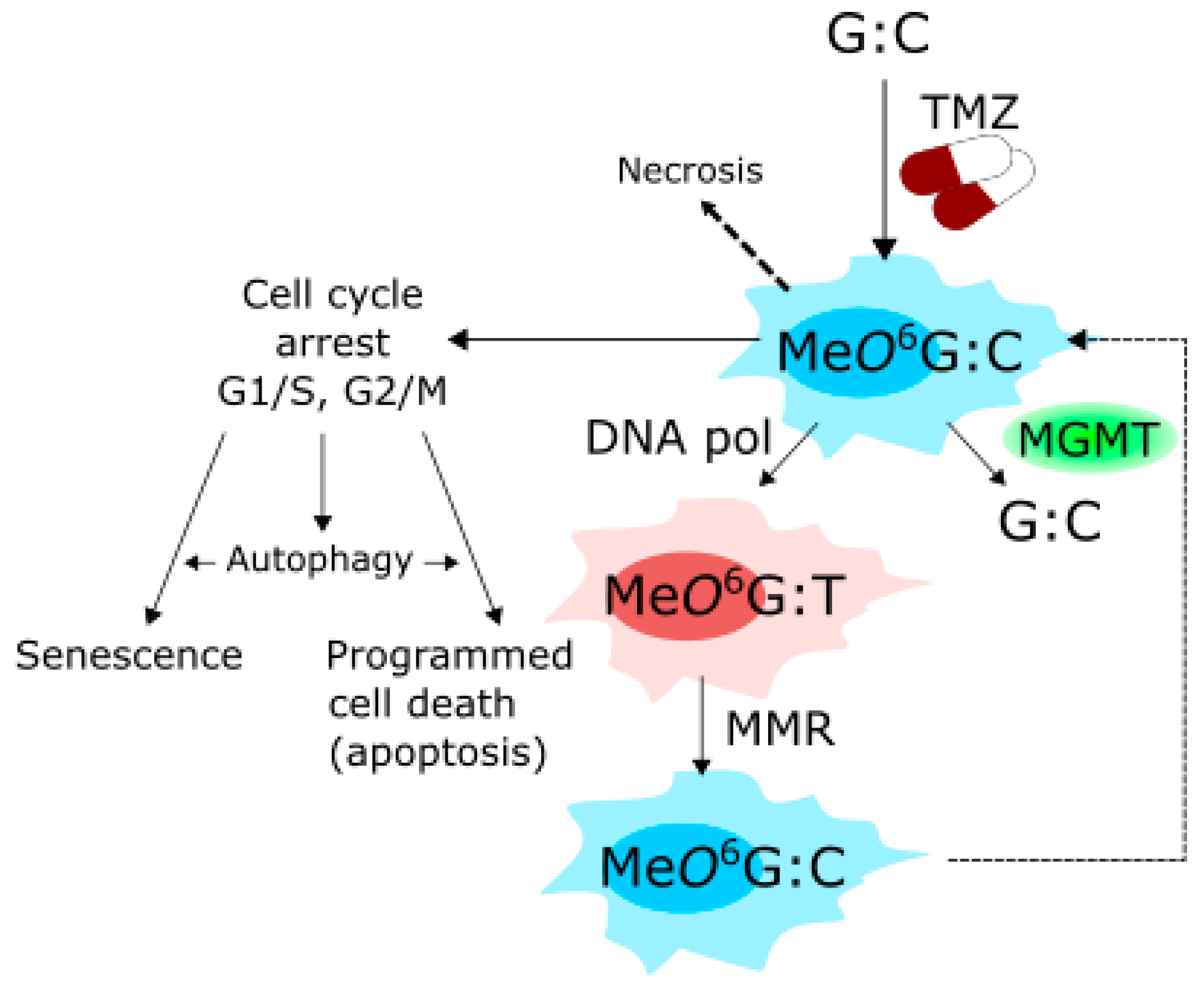
5. Therapeutic Potential
6. Conclusions and Perspectives
Author Contributions
Conflicts of Interest
References
- Seton-Rogers, S. Genomic instability: The sting of metastasis. Nat. Rev. Cancer 2018, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Bourgeais, J.; Ishac, N.; Medrzycki, M.; Brachet-Botineau, M.; Desbourdes, L.; Gouilleux-Gruart, V.; Pecnard, E.; Rouleux-Bonnin, F.; Gyan, E.; Domenech, J.; et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget 2017, 8, 41876–41889. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.B.; McNeely, S.C.; Beckmann, R.P. Achieving Precision Death with Cell-Cycle Inhibitors that Target DNA Replication and Repair. Clin. Cancer Res. 2017, 23, 3232–3240. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R. Senotherapy: Growing old and staying young? Pflugers Arch. 2017, 469, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, K.Y.; Park, S.G.; Yu, S.N.; Kim, Y.W.; Nam, H.W.; An, H.H.; Kim, Y.W.; Ahn, S.C. Mitochondrial ROS activates ERK/autophagy pathway as a protected mechanism against deoxypodophyllotoxin-induced apoptosis. Oncotarget 2017, 8, 111581–111596. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Takeuchi, T.; Kuwano, H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 2009, 16, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Hu, X.; Wu, B.; Jiang, H.; Young, C.Y.; Pang, Y.; Yuan, H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011, 307, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, W.K.; Sun, B.; Cui, M.; Liu, S.; Gao, J.; Lou, H. Dihydroptychantol A, a macrocyclic bisbibenzyl derivative, induces autophagy and following apoptosis associated with p53 pathway in human osteosarcoma U2OS cells. Toxicol. Appl. Pharmacol. 2011, 251, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S. Biological Roles of Alternative Autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar] [PubMed]
- Shen, S.; Kepp, O.; Michaud, M.; Martins, I.; Minoux, H.; Metivier, D.; Maiuri, M.C.; Kroemer, R.T.; Kroemer, G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011, 30, 4544–4556. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Vicencio, J.M.; Kepp, O.; Joza, N.; Tajeddine, N.; Kroemer, G. Life, death and burial: Multifaceted impact of autophagy. Biochem. Soc. Trans. 2008, 36 Pt 5, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, G.; Zhou, Y.; Chen, Y.; Ouyang, L.; Liu, B. Mechanisms of autophagy and relevant small-molecule compounds for targeted cancer therapy. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Jung, J.U. Autophagy genes as tumor suppressors. Curr. Opin. Cell Biol. 2010, 22, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, M.; Umemura, A.; Itoh, Y. Current status and future prospects of chemotherapy for advanced hepatocellular carcinoma. Clin. J. Gastroenterol. 2016, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.M.; Nabavi, A.; Mehdorn, H.M.; Blömer, U. Glioblastoma multiforme—Report of 267 cases treated at a single institution. Surg. Neurol. 2005, 63, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain Sci. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H. Genetic pathways to glioblastomas. Neuropathology 2005, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.M.; Kang, S.H.; Park, C.K.; Jung, S.; Park, E.S.; Lee, J.S.; Kim, S.H.; Woo, H.G. Recurrent Glioblastomas Reveal Molecular Subtypes Associated with Mechanistic Implications of Drug-Resistance. PLoS ONE 2015, 10, e0140528. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [PubMed]
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Wait, S.D.; Prabhu, R.S.; Burri, S.H.; Atkins, T.G.; Asher, A.L. Polymeric drug delivery for the treatment of glioblastoma. Neuro Oncol. 2015, 17 (Suppl. S2), ii9–ii23. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, K.; Clavreul, A.; Lagarce, F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 2015, 20, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J. Clin. Oncol. 2007, 25, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes, J.P.; Passos, J.F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 2018, 170, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Rincheval, V.; Renaud, F.; Lemaire, C.; Godefroy, N.; Trotot, P.; Boulo, V.; Mignotte, B.; Vayssière, J.-L. Bcl-2 can promote p53-dependent senescence versus apoptosis without affecting the G1/S transition. Biochem. Biophys. Res. Commun. 2002, 298, 282–288. [Google Scholar] [CrossRef]
- Goligorsky, M.S.; Hirschi, K. Stress-Induced Premature Senescence of Endothelial and Endothelial Progenitor Cells. Adv. Pharmacol. 2016, 77, 281–306. [Google Scholar] [PubMed]
- Bernal, A.; Tusell, L. Telomeres: Implications for Cancer Development. Int. J. Mol. Sci. 2018, 19, 294. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Giardini, M.A.; Segatto, M.; da Silva, M.S.; Nunes, V.S.; Cano, M.I.N. Chapter One—Telomere and Telomerase Biology. Prog. Mol. Biol. Transl. Sci. 2014, 125, 1–40. [Google Scholar] [PubMed]
- Franceschi, C.; Salvioli, S.; Garagnani, P.; de Eguileor, M.; Monti, D.; Capri, M. Immunobiography and the Heterogeneity of Immune Responses in the Elderly: A Focus on Inflammaging and Trained Immunity. Front. Immunol. 2017, 8, 982. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Dumont, P.; Dierick, J.F.; Pascal, T.; Frippiat, C.; Chainiaux, F.; Sluse, F.; Eliaers, F.; Remacle, J. Stress-induced premature senescence. Essence of life, evolution, stress, and aging. Ann. N. Y. Acad. Sci. 2000, 908, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.H.; Cameron-Smith, D.; Wessner, B.; Franzke, B. Biomarkers of Aging: From Function to Molecular Biology. Nutrients 2016, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Mikula-Pietrasik, J.; Stryczynski, L.; Uruski, P.; Tykarski, A.; Ksiazek, K. Procancerogenic activity of senescent cells: A case of the peritoneal mesothelium. Ageing Res. Rev. 2018, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Gont, A.; Perkins, T.J.; Hanson, J.E.L.; Lorimer, I.A.J. Induction of senescence in primary glioblastoma cells by serum and TGFbeta. Sci. Rep. 2017, 7, 2156. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Narita, Y.; Takami, H.; Fukushima, S.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Shibui, S.; Ichimura, K. TERT promoter mutations rather than methylation are the main mechanism for TERT upregulation in adult gliomas. Acta Neuropathol. 2013, 126, 939–941. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, M.N.; Armstrong, G.N.; Gramatges, M.M.; Bertuch, A.A.; Jhangiani, S.N.; Doddapaneni, H.; Lewis, L.; Tombrello, J.; Tsavachidis, S.; Liu, Y.; et al. Germline mutations in shelterin complex genes are associated with familial glioma. J. Natl. Cancer Inst. 2015, 107, 384. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.Y.; Kim, J.K.; Ham, S.W.; Oh, S.Y.; Kim, J.; Park, J.B.; Lee, J.Y.; Kim, S.C.; Kim, H. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumour Biol. 2016, 37, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Paget, J.A.; Restall, I.J.; Daneshmand, M.; Mersereau, J.A.; Simard, M.A.; Parolin, D.A.; Lavictoire, S.J.; Amin, M.S.; Islam, S.; Lorimer, I.A. Repression of cancer cell senescence by PKCiota. Oncogene 2012, 31, 3584–3596. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peres, E.A.; Gerault, A.N.; Valable, S.; Roussel, S.; Toutain, J.; Divoux, D.; Guillamo, J.S.; Sanson, M.; Bernaudin, M.; Petit, E. Silencing erythropoietin receptor on glioma cells reinforces efficacy of temozolomide and X-rays through senescence and mitotic catastrophe. Oncotarget 2015, 6, 2101–2119. [Google Scholar] [CrossRef] [PubMed]
- Restall, I.J.; Parolin, D.A.; Daneshmand, M.; Hanson, J.E.; Simard, M.A.; Fitzpatrick, M.E.; Kumar, R.; Lavictoire, S.J.; Lorimer, I.A. PKCiota depletion initiates mitotic slippage-induced senescence in glioblastoma. Cell Cycle 2015, 14, 2938–2948. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, H.; Wu, A. FOXO1 is crucial in glioblastoma cell tumorigenesis and regulates the expression of SIRT1 to suppress senescence in the brain. Mol. Med. Rep. 2018, 17, 2535–2542. [Google Scholar] [PubMed]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell. Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Li, X.; Che, X.; Wei, C.; He, S.; Lu, J.; Jia, Z.; Pang, K.; Fan, L. SIRT1 is a regulator of autophagy: Implications in gastric cancer progression and treatment. FEBS Lett. 2015, 589, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, J.; Guan, F.; Song, L.; Fan, R.; Zhu, H.; Hu, X.; Shen, E.; Yang, B. Copper induces cellular senescence in human glioblastoma multiforme cells through downregulation of Bmi-1. Oncol. Rep. 2013, 29, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Mustafi, S.B.; Saha, S.; Kumar Dhar Dwivedi, S.; Mukherjee, P.; Bhattacharya, R. Inhibition of BMI1 induces autophagy-mediated necroptosis. Autophagy 2016, 12, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.; Andrade, D.; Mehta, M.; Berry, W.; Benbrook, D.M.; Aravindan, N.; Herman, T.S.; Ramesh, R.; Munshi, A. Silencing BMI1 radiosensitizes human breast cancer cells by inducing DNA damage and autophagy. Oncol. Rep. 2017, 37, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, L.; Imbert, V.; Nebout, M.; Colosetti, P.; Neffati, Z.; Lagadec, P.; Verhoeyen, E.; Peng, C.; Duprez, E.; Legros, L.; et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015, 29, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, Y.; Cui, X.; Yasuda, T.; Wang, B.; Yu, D.; Sekine-Suzuki, E.; Nenoi, M. Arsenite induces premature senescence via p53/p21 pathway as a result of DNA damage in human malignant glioblastoma cells. BMB Rep. 2014, 47, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.B.; Lee, J.J.; Yun, H.H.; Im, C.N.; Kim, Y.S.; Ko, J.H.; Lee, J.H. 14-3-3beta Depletion Drives a Senescence Program in Glioblastoma Cells Through the ERK/SKP2/p27 Pathway. Mol. Neurobiol. 2017, 55, 1–12. [Google Scholar]
- Liu, Q.; Xu, X.; Zhao, M.; Wei, Z.; Li, X.; Zhang, X.; Liu, Z.; Gong, Y.; Shao, C. Berberine induces senescence of human glioblastoma cells by downregulating the EGFR-MEK-ERK signaling pathway. Mol. Cancer Ther. 2015, 14, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.W.; DiTacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature 2012, 485, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, B.; Xie, X.; Xiao, Y.F.; Yang, S.M.; Zhang, J.W. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol. Ther. 2015, 16, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Sivridis, E.; Mitrakas, A.; Kalamida, D.; Zois, C.E.; Haider, S.; Piperidou, C.; Pappa, A.; Gatter, K.C.; Harris, A.L.; et al. Autophagy and lysosomal related protein expression patterns in human glioblastoma. Cancer Biol. Ther. 2014, 15, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Sun, L.; Wang, C.; Huang, H.; Liu, J.; Liao, W.; Shi, M. LC3A-positive “stone-like” structures predict an adverse prognosis of gastric cancer. Anat. Rec. (Hoboken) 2014, 297, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Kloog, Y.; Pinkas-Kramarski, R. Ras and autophagy in cancer development and therapy. Oncotarget 2014, 5, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Zhang, J.; Guo, X.; Wang, J.; Li, J.; Gao, X.; Guo, X.; Li, T.; Xu, S.; Zhang, P.; et al. CREBRF is a potent tumor suppressor of glioblastoma by blocking hypoxia-induced autophagy via the CREB3/ATG5 pathway. Int. J. Oncol. 2016, 49, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed]
- Manal, M.; Chandrasekar, M.J.; Gomathi Priya, J.; Nanjan, M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef] [PubMed]
- Nervi, C.; De Marinis, E.; Codacci-Pisanelli, G. Epigenetic treatment of solid tumours: A review of clinical trials. Clin. Epigenet. 2015, 7, 127. [Google Scholar] [CrossRef] [PubMed]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. 2010, 3, ra81. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B.; Bates, P.A.; Paik, J.; Jacobs, S.C.; Lindahl, T. Repair of alkylated DNA: Recent advances. DNA Repair. (Amst) 2007, 6, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Li, L.T.; Xin, Y.; Zhang, L.; Liu, Y.Q.; Zheng, J.N. Strategies to improve the killing of tumors using temozolomide: Targeting the DNA repair protein MGMT. Curr. Med. Chem. 2012, 19, 3886–3892. [Google Scholar] [CrossRef] [PubMed]
- Strik, H.M.; Marosi, C.; Kaina, B.; Neyns, B. Temozolomide dosing regimens for glioma patients. Curr. Neurol. Neurosci. Rep. 2012, 12, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Thome, M.P.; Bueno e Silva, M.M.; Pelegrini, A.L.; Ledur, P.F.; Garicochea, B.; Zamin, L.L.; Lenz, G. Resveratrol abrogates the temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the temozolomide-induced senescence in glioma cells. BMC Cancer 2013, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yang, X.J.; Zhang, W.G.; Ji, Y.W.; Pan, Q. Mechanism of thalidomide to enhance cytotoxicity of temozolomide in U251-MG glioma cells in vitro. Chin. Med. J. (Engl.) 2009, 122, 1260–1266. [Google Scholar] [PubMed]
- Filippi-Chiela, E.C.; Bueno e Silva, M.M.; Thome, M.P.; Lenz, G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 2015, 11, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhang, Y.; Liu, Y.; Yamada, K.; Tso, J.L.; Menjivar, J.C.; Tian, J.Y.; Yong, W.H.; Schaue, D.; Mischel, P.S.; et al. Protective properties of radio-chemoresistant glioblastoma stem cell clones are associated with metabolic adaptation to reduced glucose dependence. PLoS ONE 2013, 8, e80397. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [PubMed]
- Poklepovic, A.; Gewirtz, D.A. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014, 10, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, H.K.; Lee, N.H.; Yi, H.Y.; Kim, H.S.; Hong, S.H.; Hong, Y.K.; Joe, Y.A. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015, 360, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.W.; Jeon, H.Y.; Kim, H. Verapamil augments carmustine- and irradiation-induced senescence in glioma cells by reducing intracellular reactive oxygen species and calcium ion levels. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Foreman, P.M.; Friedman, G.K.; Cassady, K.A.; Markert, J.M. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017, 14, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Panek, W.K.; Kane, J.R.; Young, J.S.; Rashidi, A.; Kim, J.W.; Kanojia, D.; Lesniak, M.S. Hitting the nail on the head: Combining oncolytic adenovirus-mediated virotherapy and immunomodulation for the treatment of glioma. Oncotarget 2017, 8, 89391–89405. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Hong, J.; Kwon, O.J.; Yun, C.O. A hypoxia- and telomerase-responsive oncolytic adenovirus expressing secretable trimeric TRAIL triggers tumour-specific apoptosis and promotes viral dispersion in TRAIL-resistant glioblastoma. Sci. Rep. 2018, 8, 1420. [Google Scholar] [CrossRef] [PubMed]
- Proske, J.; Walter, L.; Bumes, E.; Hutterer, M.; Vollmann-Zwerenz, A.; Eyupoglu, I.Y.; Savaskan, N.E.; Seliger, C.; Hau, P.; Uhl, M. Adaptive Immune Response to and Survival Effect of Temozolomide- and Valproic Acid-induced Autophagy in Glioblastoma. Anticancer Res. 2016, 36, 899–905. [Google Scholar] [PubMed]
- Lefranc, F.; Kiss, R. Autophagy, the Trojan horse to combat glioblastomas. Neurosurg. Focus 2006, 20, E7. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic autophagy in cancer therapy. Int. J. Mol. Sci. 2014, 15, 10034–10051. [Google Scholar] [CrossRef] [PubMed]
- Nanegrungsunk, D.; Onchan, W.; Chattipakorn, N.; Chattipakorn, S.C. Current evidence of temozolomide and bevacizumab in treatment of gliomas. Neurol. Res. 2015, 37, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Piao, Y.; Henry, V.; Tiao, N.; de Groot, J.F. Interferon-regulatory factor-1 (IRF1) regulates bevacizumab induced autophagy. Oncotarget 2015, 6, 31479–31492. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Li, D.; Sun, K.; Wang, J.; Liu, Y.; Song, J.R.; Zhao, Q.D.; Zhang, S.S.; Deng, W.J.; Zhao, X.; et al. Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J. Mol. Med. (Berl.) 2013, 91, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, J.; Liu, Z.; Pan, L.; Xu, G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol. Lett. 2018, 15, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Cardoso-Carneiro, D.; Valbom, I.; Cury, F.P.; Silva, V.A.; Granja, S.; Reis, R.M.; Baltazar, F.; Martinho, O. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017, 8, 103657–103670. [Google Scholar] [CrossRef] [PubMed]
- Muller-Greven, G.; Carlin, C.R.; Burgett, M.E.; Ahluwalia, M.S.; Lauko, A.; Nowacki, A.S.; Herting, C.J.; Qadan, M.A.; Bredel, M.; Toms, S.A.; et al. Macropinocytosis of Bevacizumab by glioblastoma cells in the perivascular niche affects their survival. Clin. Cancer Res. 2017, 23, 7059–7071. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.; Caire, F.; Guyotat, J.; Menei, P.; Metellus, P.; Pallud, J. Carmustine wafer implantation for high-grade gliomas: Evidence-based safety efficacy and practical recommendations from the Neuro-oncology Club of the French Society of Neurosurgery. Neurochirurgie 2017, 63, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Roos, W.P.; Kramer, O.H.; Strik, H.M.; Kaina, B. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim. Biophys. Acta 1868, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Ray, S.K. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: Overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis 2016, 21, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Clark, A.J.; Chan, D.C.; Ware, J.L.; Holt, S.E.; Chidambaram, A.; Fillmore, H.L.; Broaddus, W.C. Wilms’ tumor 1 silencing decreases the viability and chemoresistance of glioblastoma cells in vitro: A potential role for IGF-1R de-repression. J. Neuroncol. 2011, 103, 87–102. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlowska, E.; Szczepanska, J.; Szatkowska, M.; Blasiak, J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective. Int. J. Mol. Sci. 2018, 19, 889. https://doi.org/10.3390/ijms19030889
Pawlowska E, Szczepanska J, Szatkowska M, Blasiak J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective. International Journal of Molecular Sciences. 2018; 19(3):889. https://doi.org/10.3390/ijms19030889
Chicago/Turabian StylePawlowska, Elzbieta, Joanna Szczepanska, Magdalena Szatkowska, and Janusz Blasiak. 2018. "An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective" International Journal of Molecular Sciences 19, no. 3: 889. https://doi.org/10.3390/ijms19030889
APA StylePawlowska, E., Szczepanska, J., Szatkowska, M., & Blasiak, J. (2018). An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective. International Journal of Molecular Sciences, 19(3), 889. https://doi.org/10.3390/ijms19030889