Role of Non-Myocyte Gap Junctions and Connexin Hemichannels in Cardiovascular Health and Disease: Novel Therapeutic Targets?


Abstract

1. Introduction
2. Connexins and Cardiac Fibroblasts
2.1. Fibroblast Identification and Function
2.2. Fibroblast Connexins and Cell-Cell Coupling
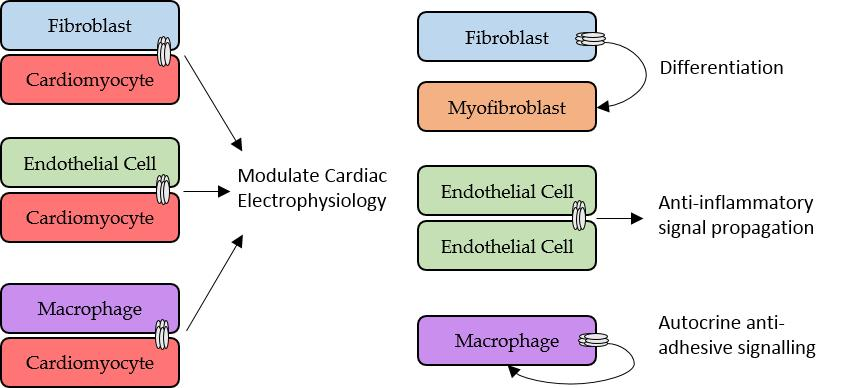
2.3. Role of Fibroblasts in Cardiac Electrophysiology
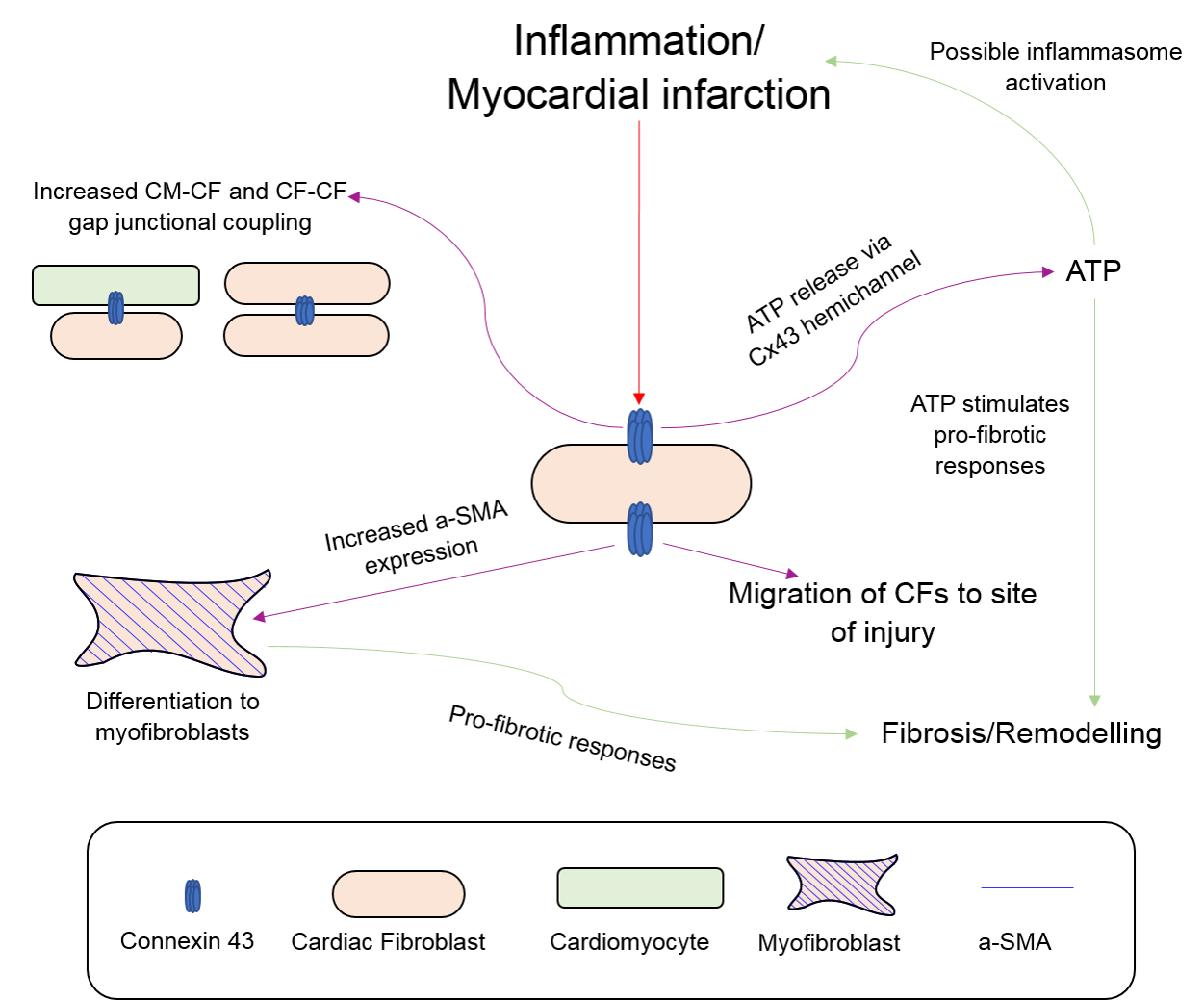
2.4. Fibroblasts, Connexins and Myocardial Remodelling
3. Connexins and Endothelial Cells
3.1. Roles of Endothelial Cells in Cardiac Muscle and the Vasculature
3.2. Endothelial Cell-Cardiomyocyte Gap Junctional Signalling
3.3. Endothelial Cells, Connexins, and Vasomotor Control
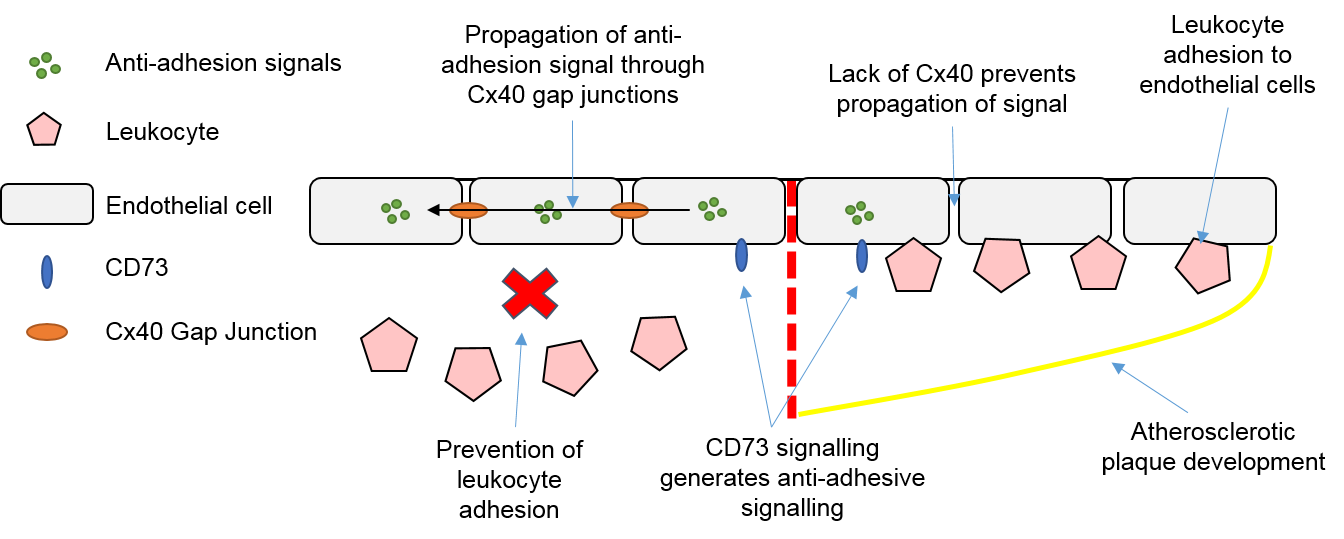
3.4. Endothelial Cells, Connexins and Atherosclerosis
4. Connexins and Macrophages
4.1. Roles of Macrophages in the Heart and Vasculature
4.2. Macrophages, Connexins and Electrical Conduction in the Heart
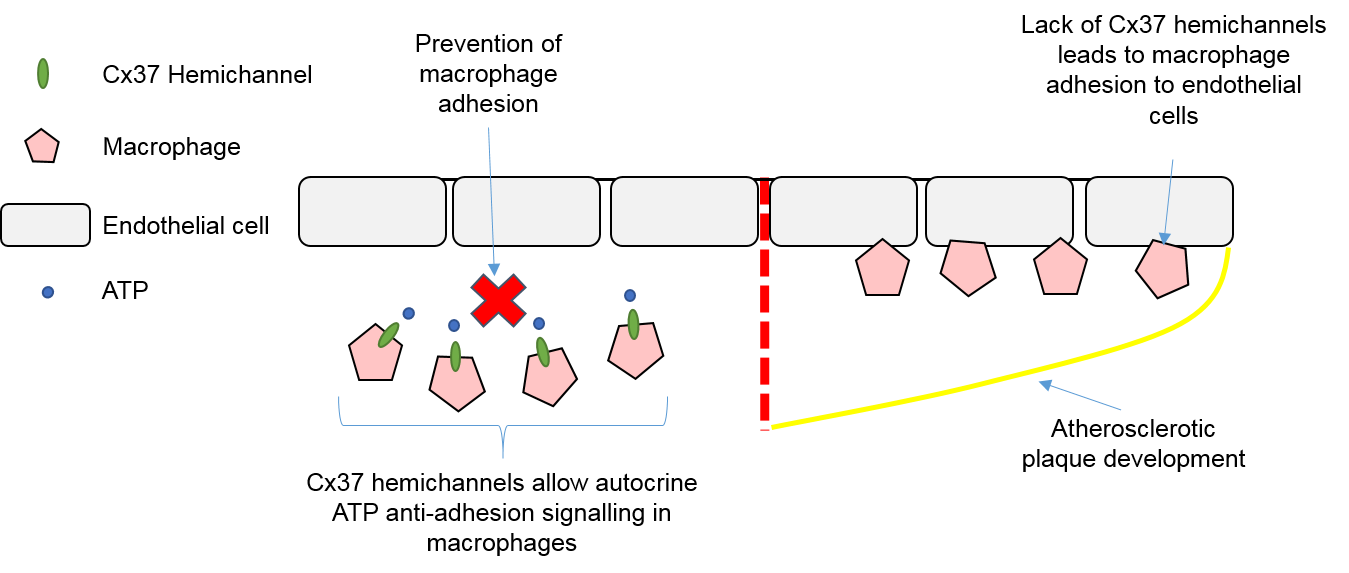
4.3. Macrophages, Connexins, and Atherosclerosis
5. Non-Myocyte Gap Junctions and Hemichannels as Novel Therapeutic Targets
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CMs | Cardiomyocytes |
| CFs | Cardiac Fibroblasts |
| ECs | Endothelial Cells |
| SMCs | Smooth Muscle Cells |
| ECM | Extracellular Matrix |
| Cx | Connexin |
| SAN | Sinoatrial Node |
| AVN | Atrioventricular Node |
| Na+ | Sodium |
| K+ | Potassium |
| ATP | Adenosine triphosphate |
| Ca2+ | Calcium |
| DDR2 | Discoidin Domain Receptor 2 |
| POSTN | Periostin |
| FSP1 | Fibroblast Specific Protein 1 |
| α-SMA | α-Smooth Muscle Actin |
| MI | Myocardial Infarction |
| Cx43FSP1KO | Fibroblast-Specific Protein-1 Driven Conditional Cx43 Knockout |
| TGF-β | Transforming Growth Factor-β |
| NO | Nitric Oxide |
| ET-1 | Endothelin-1 |
| eNOS | Endothelial Nitric Oxide Synthase |
| VEGF | Vascular Endothelial Growth Factor |
| HHcy | Hyperhomocysteinemia |
| Hcy | Homocysteine |
| HUVECs | Human Umbilical Vein Endothelial Cells |
| VCAM-1 | Vascular Cell Adhesion Molecule-1 |
| IL | Interleukin |
| MCP-1 | Monocyte Chemotactic Protein-1 |
| TNF-α | Tumour Necrosis Factor-α |
| LPS | Lipopolysaccharide |
| RhoA | Ras homolog gene family, member A |
| HLSS | High laminar shear stress |
| LDL | Low-Density Lipoprotein |
| DAMPs | Danger-associated Molecular Patterns |
| NOD | Nucleotide Oligomerisation Domain |
| NLRP3 | NOD-like Receptor Protein-3 |
| αCT1 | α-Connexin Carboxyl-Terminus |
| ZO-1 | Zonula Occludens-1 |
| CT | Carboxyl-Terminus |
| AF | Atrial Fibrillation |
| PDGFR | Platelet Derived Growth Factor Receptor |
| GTN | Glyceryl Trinitrate |
| JM2 | Juxtamembrane 2 |
References
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, E.A.; Matkovich, S.J. Cardiomyocytes structure, function and associated pathologies. Int. J. Biochem. Cell Biol. 2005, 37, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Lajiness, J.D.; Conway, S.J. The Dynamic Role of Cardiac Fibroblasts in Development and Disease. J. Cardiovasc. Transl. Res. 2012, 5, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 2017, 9 (Suppl. 1), S30–S35. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An Abundant Tissue Macrophage Population in the Adult Murine Heart with a Distinct Alternatively-Activated Macrophage Profile. PLoS ONE 2012, 7, e36814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, W.T. Recounting cardiac cellular composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Pontén, A.; Fleischmann, B.K.; Jovinge, S. Cardiomyocyte cell cycle control and growth estimation in vivo—An analysis based on cardiomyocyte nuclei. Cardiovasc. Res. 2010, 86, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1883–H1891. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; Antoni, M.; Debuque, R.J.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2015. [Google Scholar] [CrossRef]
- Sheikh, F.; Ross, R.S.; Chen, J. Cell-Cell Connection to Cardiac Disease. Trends Cardiovasc. Med. 2009, 19, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.A.; van Veen, T.A.B.; de Bakker, J.M.T.; van Rijen, H.V.M. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A.; Paul, D.L. Gap Junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.W. Role of Gap Junctions in Cardiac Conduction and Development. Circ. Res. 2000, 87, 346. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Desplantez, T. Cardiac Cx43, Cx40 and Cx45 co-assembling: Involvement of connexins epitopes in formation of hemichannels and Gap junction channels. BMC Cell Biol. 2017, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Kostin, S.; Rieger, M.; Dammer, S.; Hein, S.; Richter, M.; Klovekorn, W.P.; Bauer, E.P.; Schaper, J. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol. Cell. Biochem. 2003, 242, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Dupont, E.; Matsushita, T.; Kaba, R.A.; Vozzi, C.; Coppen, S.R.; Khan, N.; Kaprielian, R.; Yacoub, M.H.; Severs, N.J. Altered connexin expression in human congestive heart failure. J. Mol. Cell. Cardiol. 2001, 33, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gutstein, D.E.; Morley, G.E.; Tamaddon, H.; Vaidya, D.; Schneider, M.D.; Chen, J.; Chien, K.R.; Stuhlmann, H.; Fishman, G.I. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 2001, 88, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 2000, 101, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kleber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Gollob, M.H.; Jones, D.L.; Krahn, A.D.; Danis, L.; Gong, X.Q.; Shao, Q.; Liu, X.; Veinot, J.P.; Tang, A.S.; Stewart, A.F.; et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 2006, 354, 2677–2688. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Zhang, X.L.; Wang, X.H.; Tan, H.W.; Shi, H.F.; Jiang, W.F.; Fang, W.Y.; Liu, X. Connexin40 nonsense mutation in familial atrial fibrillation. Int. J. Mol. Med. 2010, 26, 605–610. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, Y.Q.; Liu, X.; Zhang, X.L.; Wang, X.H.; Tan, H.W.; Shi, H.F.; Jiang, W.F.; Fang, W.Y. Novel connexin40 missense mutations in patients with familial atrial fibrillation. EP Europace 2010, 12, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Lubkemeier, I.; Andrie, R.; Lickfett, L.; Bosen, F.; Stockigt, F.; Dobrowolski, R.; Draffehn, A.M.; Fregeac, J.; Schultze, J.L.; Bukauskas, F.F.; et al. The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J. Mol. Cell. Cardiol. 2013, 65, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Kumai, M.; Nishii, K.; Nakamura, K.; Takeda, N.; Suzuki, M.; Shibata, Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 2000, 127, 3501–3512. [Google Scholar] [PubMed]
- Frank, M.; Wirth, A.; Andrie, R.P.; Kreuzberg, M.M.; Dobrowolski, R.; Seifert, G.; Offermanns, S.; Nickenig, G.; Willecke, K.; Schrickel, J.W. Connexin45 provides optimal atrioventricular nodal conduction in the adult mouse heart. Circ. Res. 2012, 111, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hills, M.D.; Ye, W.G.; Tong, X.; Bai, D. Atrial Fibrillation-Linked Germline GJA5/Connexin40 Mutants Showed an Increased Hemichannel Function. PLoS ONE 2014, 9, e95125. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patel, D.; Gemel, J.; Xu, Q.; Simon, A.R.; Lin, X.; Matiukas, A.; Beyer, E.C.; Veenstra, R.D. Atrial fibrillation-associated Connexin40 mutants make hemichannels and synergistically form gap junction channels with novel properties. FEBS Lett. 2014, 588, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.A.; Bowers, S.L.K.; Baudino, T.A. Cardiac Fibroblast: The Renaissance Cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Norris, R.A.; Kohl, P.; Markwald, R. The Living Scar—Cardiac Fibroblasts and the Injured Heart. Trends Mol. Med. 2016, 22, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Gourdie, R.G.; Dimmeler, S.; Kohl, P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 2016, 15, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and Myofibroblasts: What Are We Talking About? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; Carver, W.; Giles, W.; Borg, T.K. Cardiac fibroblasts: Friend or foe? Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1015–H1026. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.; Ubil, E. Cardiac Fibroblast in Development and Wound Healing. J. Mol. Cell. Cardiol. 2014, 70, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—From repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Louault, C.; Benamer, N.; Faivre, J.-F.; Potreau, D.; Bescond, J. Implication of connexins 40 and 43 in functional coupling between mouse cardiac fibroblasts in primary culture. Biochim. Biophys. Acta Biomembr. 2008, 1778, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Gaudesius, G.; Miragoli, M.; Thomas, S.P.; Rohr, S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ. Res. 2003, 93, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Kohl, P.; Camelliti, P. Fibroblast-myocyte connections in the heart. Heart Rhythm 2012, 9, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Green, C.R.; LeGrice, I.; Kohl, P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004, 94, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, A.M.; Camelliti, P.; Walker, N.L.; Burton, F.L.; Cobbe, S.M.; Kohl, P.; Smith, G.L. Prolongation of atrio-ventricular node conduction in a rabbit model of ischaemic cardiomyopathy: Role of fibrosis and connexin remodelling. J. Mol. Cell. Cardiol. 2016, 94, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Devlin, G.P.; Matthews, K.G.; Kohl, P.; Green, C.R. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc. Res. 2004, 62, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, C.; Mohandas, P.; Louie, K.L.; Benamer, N.; Bapat, A.C.; Morley, G.E. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 2010, 107, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.A.; Camelliti, P.; Rog-Zielinska, E.A.; Siedlecka, U.; Poggioli, T.; O’Toole, E.T.; Knöpfel, T.; Kohl, P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. USA 2016, 113, 14852–14857. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.L.; Burton, F.L.; Kettlewell, S.; Smith, G.L.; Cobbe, S.M. Mapping of epicardial activation in a rabbit model of chronic myocardial infarction. J. Cardiovasc. Electrophysiol. 2007, 18, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Kohl, P.; Camelliti, P.; Burton, F.L.; Smith, G.L. Electrical coupling of fibroblasts and myocytes: Relevance for cardiac propagation. J. Electrocardiol. 2005, 38 (Suppl. 4), 45–50. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, W.G. Ventricular Scars and Ventricular Tachycardia. Trans. Am. Clin. Climatol. Assoc. 2009, 120, 403–412. [Google Scholar] [PubMed]
- Morita, N.; Mandel, W.J.; Kobayashi, Y.; Karagueuzian, H.S. Cardiac fibrosis as a determinant of ventricular tachyarrhythmias. J. Arrhythm. 2014, 30, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.B.; Calloe, K. Human atrial fibroblasts and their contribution to supraventricular arrhythmia. Physiol. Rep. 2016, 4, e12711. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kohl, P.; Gourdie, R.G. Fibroblast-myocyte electrotonic coupling: Does it occur in native cardiac tissue? J. Mol. Cell. Cardiol. 2014, 70, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Gaudesius, G.; Rohr, S. Electrotonic Modulation of Cardiac Impulse Conduction by Myofibroblasts. Circ. Res. 2006, 98, 801. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Salvarani, N.; Rohr, S. Myofibroblasts Induce Ectopic Activity in Cardiac Tissue. Circ. Res. 2007, 101, 755. [Google Scholar] [CrossRef] [PubMed]
- Askar, S.F.; Bingen, B.O.; Swildens, J.; Ypey, D.L.; van der Laarse, A.; Atsma, D.E.; Zeppenfeld, K.; Schalij, M.J.; de Vries, A.A.; Pijnappels, D.A. Connexin43 silencing in myofibroblasts prevents arrhythmias in myocardial cultures: Role of maximal diastolic potential. Cardiovasc. Res. 2012, 93, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Zlochiver, S.; Muñoz, V.; Vikstrom, K.L.; Taffet, S.M.; Berenfeld, O.; Jalife, J. Electrotonic Myofibroblast-to-Myocyte Coupling Increases Propensity to Reentrant Arrhythmias in Two-Dimensional Cardiac Monolayers. Biophys. J. 2008, 95, 4469–4480. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, V.M.; Mezzano, V.; Mirams, G.R.; Maass, K.; Li, Z.; Cerrone, M.; Vasquez, C.; Bapat, A.; Delmar, M.; Morley, G.E. Connexin43 contributes to electrotonic conduction across scar tissue in the intact heart. Sci. Rep. 2016, 6, 26744. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Xie, J.; Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2011, 89, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Jin, J.; Shen, W.; Tsui, H.; Shi, Y.; Wang, Y.; Zhang, Y.; Hao, G.; Wu, J.; Chen, S.; et al. Mkk4 Is a Negative Regulator of the Transforming Growth Factor Beta 1 Signaling Associated With Atrial Remodeling and Arrhythmogenesis With Age. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2014, 3, e000340. [Google Scholar] [CrossRef] [PubMed]
- Ashihara, T.; Haraguchi, R.; Nakazawa, K.; Namba, T.; Ikeda, T.; Nakazawa, Y.; Ozawa, T.; Ito, M.; Horie, M.; Trayanova, N.A. The Role of Fibroblasts in Complex Fractionated Electrograms During Persistent/Permanent Atrial Fibrillation: Implications for Electrogram-Based Catheter Ablation. Circ. Res. 2012, 110, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Xia, L. Excitation-Contraction Coupling between Human Atrial Myocytes with Fibroblasts and Stretch Activated Channel Current: A Simulation Study. Comput. Math. Methods Med. 2013, 2013, 238676. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Qi, X.Y.; Huang, H.; Nattel, S. Fibroblast Electrical Remodeling in Heart Failure and Potential Effects on Atrial Fibrillation. Biophys. J. 2014, 107, 2444–2455. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bunch, T.J.; Hohnloser, S.H.; Gersh, B.J. Mechanisms of Sudden Cardiac Death in Myocardial Infarction Survivors. Circulation 2007, 115, 2451. [Google Scholar] [CrossRef] [PubMed]
- Asazuma-Nakamura, Y.; Dai, P.; Harada, Y.; Jiang, Y.; Hamaoka, K.; Takamatsu, T. Cx43 contributes to TGF-β signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Exp. Cell Res. 2009, 315, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-β Signaling in Myocardial Infarction and Cardiac Remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Kovacs, A.; Kanter, E.M.; Yamada, K.A. Reduced expression of Cx43 attenuates ventricular remodeling after myocardial infarction via impaired TGF-β signaling. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H477–H487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kanter, E.M.; Laing, J.G.; Aprhys, C.; Johns, D.C.; Kardami, E.; Yamada, K.A. Connexin43 Expression Levels Influence Intercellular Coupling and Cell Proliferation of Native Murine Cardiac Fibroblasts. Cell Commun. Adhes. 2008, 15, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Naranjo, A.; Cormie, P.; Serrano, A.E.; Wang, C.M.; Thrasivoulou, C.; Sutcliffe, J.E.; Gilmartin, D.J.; Tsui, J.; Serena, T.E.; Phillips, A.R.; et al. Overexpression of the gap junction protein Cx43 as found in diabetic foot ulcers can retard fibroblast migration. Cell Biol. Int. 2012, 36, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.S.; van Steensel, M.A.; Hodgins, M.B.; Martin, P.E. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen. 2009, 17, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Mori, R.; Power, K.T.; Wang, C.M.; Martin, P.; Becker, D.L. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 2006, 119 Pt 24, 5193–5203. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.S.; Pollok, S.; Flint, D.J.; Brandner, J.M.; Martin, P.E. The connexin mimetic peptide Gap27 increases human dermal fibroblast migration in hyperglycemic and hyperinsulinemic conditions in vitro. J. Cell. Physiol. 2012, 227, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ongstad, E.L.; Gourdie, R.G. Abstract 14887: A Connexin43 CT-Mimetic Peptide Increases Fibroblast Migration in an in vitro Model of the Injury Border Zone. Circulation 2013, 128 (Suppl. 22), A14887. [Google Scholar]
- Obert, E.; Strauss, R.; Brandon, C.; Grek, C.; Ghatnekar, G.; Gourdie, R.; Rohrer, B. Targeting the tight junction protein, zonula occludens-1, with the connexin43 mimetic peptide, αCT1, reduces VEGF-dependent RPE pathophysiology. J. Mol. Med. 2017, 95, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin 43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflüg. Arch. Eur. J. Physiol. 2010, 460, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.O. Ischemia Induces Closure of Gap Junctional Channels and Opening of Hemichannels in Heart-derived Cells and Tissue. Cell. Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Soleymani, S.; Madakshire, R.; Insel, P.A. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y(2) receptors. FASEB J. 2012, 26, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The importance of NLRP3 inflammasome in heart failure. J. Card. Fail. 2015, 21, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Green, C.R.; Kho, D.T.; Zhang, J.; Graham, E.S.; Acosta, M.L.; Rupenthal, I.D. The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 Protein Induces Reactive Oxygen Species to Promote Smad Protein Signaling and Fibrosis Independent from the Inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, O.; Ranheim, T.; Vinge, L.E.; Bliksoen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kanter, E.M.; Yamada, K.A. Remodeling of Cardiac Fibroblasts Following Myocardial Infarction Results in Increased Gap Junction Intercellular Communication. Cardiovasc. Pathol. 2010, 19, e233–e240. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.C.H.; Davis, M.E.; Lisowski, L.K.; Lee, R.T. Endothelial-Cardiomyocyte Interactions in Cardiac Development and Repair. Ann. Rev. Physiol. 2006, 68, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Fountoulaki, K.; Dagres, N.; Iliodromitis, E.K. Cellular Communications in the Heart. Card. Fail. Rev. 2015, 1, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.C.S.V.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Day, K.H.; Damon, D.N.; Duling, B.R. Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 9989–9994. [Google Scholar] [CrossRef] [PubMed]
- Narmoneva, D.A.; Vukmirovic, R.; Davis, M.E.; Kamm, R.D.; Lee, R.T. Endothelial Cells Promote Cardiac Myocyte Survival and Spatial Reorganization: Implications for Cardiac Regeneration. Circulation 2004, 110, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Givvimani, S.; Qipshidze, N.; Tyagi, N.; Mishra, P.K.; Sen, U.; Tyagi, S.C. Synergism between arrhythmia and hyperhomo-cysteinemia in structural heart disease. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 107–119. [Google Scholar] [PubMed]
- Morel, S.; Braunersreuther, V.; Chanson, M.; Bouis, D.; Rochemont, V.; Foglia, B.; Pelli, G.; Sutter, E.; Pinsky, D.J.; Mach, F.; et al. Endothelial Cx40 limits myocardial ischaemia/reperfusion injury in mice. Cardiovasc. Res. 2014, 102, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Chadjichristos, C.E.; Scheckenbach, K.E.L.; van Veen, T.A.B.; Richani Sarieddine, M.Z.; de Wit, C.; Yang, Z.; Roth, I.; Bacchetta, M.; Viswambharan, H.; Foglia, B.; et al. Endothelial-Specific Deletion of Connexin40 Promotes Atherosclerosis by Increasing CD73-Dependent Leukocyte Adhesion. Circulation 2010, 121, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Angelov, S.N.; Simon, A.M.; Burt, J.M. Cx40 Is Required for, and Cx37 Limits, Postischemic Hindlimb Perfusion, Survival and Recovery. J. Vasc. Res. 2011, 49, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Angelov, S.N.; Simon, A.M.; Burt, J.M. Compromised regulation of tissue perfusion and arteriogenesis limit, in an AT1R-independent fashion, recovery of ischemic tissue in Cx40−/− mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H816–H827. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, C.; Ziegelhöffer, B.; Kostelka, M.; Stepan, H.; Mohr, F.-W.; Dhein, S. Knock-down of endothelial connexins impairs angiogenesis. Pharmacol. Res. 2012, 65, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Pogoda, K.; Ritter, A.; Münzing, S.; Pohl, U. Gap junctional communication controls the overall endothelial calcium response to vasoactive agonists. Cardiovasc. Res. 2012, 93, 508–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pfenniger, A.; Derouette, J.-P.; Verma, V.; Lin, X.; Foglia, B.; Coombs, W.; Roth, I.; Satta, N.; Dunoyer-Geindre, S.; Sorgen, P.; et al. Gap Junction Protein Cx37 Interacts With Endothelial Nitric Oxide Synthase in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 827. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Boittin, F.-X.; Bény, J.-L.; Haefliger, J.-A. Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1365–H1373. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Hill, C.E. Incidence of Myoendothelial Gap Junctions in the Proximal and Distal Mesenteric Arteries of the Rat Is Suggestive of a Role in Endothelium-Derived Hyperpolarizing Factor—Mediated Responses. Circ. Res. 2000, 86, 341. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.G.; Segal, S.S. Electrical Coupling Between Endothelial Cells and Smooth Muscle Cells in Hamster Feed Arteries. Circ. Res. 2000, 87, 474. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, K.-T.; Zhao, L.; Li, X.-Z.; Zhang, Z.-S.; Shi, W.-Y.; Zhu, H.; Wei, L.-L.; Si, J.-Q. Myoendothelial coupling is unidirectional in guinea pig spiral modiolar arteries. Microvasc. Res. 2012, 84, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Wong, C.; Sutter, E.; Cuhlmann, S.; Dunoyer-Geindre, S.; Mach, F.; Horrevoets, A.J.; Evans, P.C.; Krams, R.; Kwak, B.R. Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J. Mol. Cell. Cardiol. 2012, 53, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-I.; Lu, C.-S.; Wu, Y.-J.; Chen, C.-C.; Hong, R.-C.; Ko, Y.-S.; Shiao, M.-S.; Severs, N.J.; Tsai, C.-H. Reduced Expression of Endothelial Connexin37 and Connexin40 in Hyperlipidemic Mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1391. [Google Scholar] [CrossRef] [PubMed]
- Cizek, S.M.; Bedri, S.; Talusan, P.; Silva, N.; Lee, H.; Stone, J.R. Risk Factors for Atherosclerosis and the Development of Pre-Atherosclerotic Intimal Hyperplasia. Cardiovasc. Pathol. 2007, 16, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Dubuis, C.; Lambelet, M.; Le Gal, L.; Alonso, F.; Corpataux, J.-M.; Déglise, S.; Haefliger, J.-A. Connexin37 reduces smooth muscle cell proliferation and intimal hyperplasia in a mouse model of carotid artery ligation. Cardiovasc. Res. 2017, 113, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Coon, B.G.; Gillis, N.; Chen, Z.; Qiu, J.; Chittenden, T.W.; Burt, J.M.; Schwartz, M.A.; Hirschi, K.K. Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat. Commun. 2017, 8, 2149. [Google Scholar] [CrossRef] [PubMed]
- Burt, J.M.; Nelson, T.K.; Simon, A.M.; Fang, J.S. Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1103–C1112. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.L.; Pontifex, T.K.; Li, H.; Solan, J.L.; Lampe, P.D.; Sorgen, P.L.; Burt, J.M. Regulation of Cx37 channel and growth suppressive properties by phosphorylation. J. Cell Sci. 2017, 130, 3308–3321. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Akita, N.; Yoshida, K.; Hayashi, T.; Suzuki, K. Connexin32 protects against vascular inflammation by modulating inflammatory cytokine expression by endothelial cells. Exp. Cell Res. 2011, 317, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Kawamoto, E.; Takagi, Y.; Akita, N.; Hayashi, T.; Park, E.J.; Suzuki, K.; Shimaoka, M. Gap junction-mediated regulation of endothelial cellular stiffness. Sci. Rep. 2017, 7, 6134. [Google Scholar] [CrossRef] [PubMed]
- Harari, E.; Guo, L.; Smith, S.L.; Braumann, R.E.; Virmani, R.; Finn, A.V. Heart-resident macrophages: Are they involved in the rhythm of every beat? J. Thorac. Dis. 2017, 9, 2264–2267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Swirski, F.K.; Robbins, C.S.; Nahrendorf, M. Development and Function of Arterial and Cardiac Macrophages. Trends Immunol. 2016, 37, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Nahrendorf, M. Monocytes in Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Hilhorst, M.; Harrison, D.G.; Goronzy, J.J.; Weyand, C.M. Macrophages in Vascular Inflammation—From Atherosclerosis to Vasculitis. Autoimmunity 2015, 48, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [PubMed]
- Temple, I.P.; Inada, S.; Dobrzynski, H.; Boyett, M.R. Connexins and the atrioventricular node. Heart Rhythm 2013, 10, 297–304. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, N.M.; Wang, L.; Herren, A.W.; Wang, J.; Shenasa, F.; Bers, D.M.; Lindsey, M.L.; Ripplinger, C.M. Atherosclerosis exacerbates arrhythmia following myocardial infarction: Role of myocardial inflammation. Heart Rhythm 2015, 12, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.-P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Chanson, M.; Nguyen, T.D.; Glass, A.M.; Richani Sarieddine, M.Z.; Meens, M.J.; Burnier, L.; Kwak, B.R.; Taffet, S.M. Titration of the gap junction protein Connexin43 reduces atherogenesis. Thromb. Haemost. 2014, 112, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Chanson, M.; Kwak, B.R. Connexins in atherosclerosis. Biochim. Biophys. Acta Biomembr. 2013, 1828, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.J.; Dai, S.; Gribar, S.C.; Richardson, W.; Kohler, J.W.; Hoffman, R.A.; Branca, M.F.; Li, J.; Shi, X.-H.; Sodhi, C.P.; et al. A Role for Connexin43 in Macrophage Phagocytosis and Host Survival after Bacterial Peritoneal Infection. J. Immunol. 2008, 181, 8534–8543. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, L.; Yang, J.; Li, Y.; Bai, W.; Liu, N.; Li, W.; Gao, Y.; Xu, L.; Liu, Z.; et al. LDL acts as an opsonin enhancing the phagocytosis of group A Streptococcus by monocyte and whole human blood. Med. Microbiol. Immunol. 2016, 205, 155–162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schulz, R.; Görge, P.M.; Görbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Kjølbye, A.L.; Haugan, K.; Hennan, J.K.; Petersen, J.S. Pharmacological Modulation of Gap Junction Function with the Novel Compound Rotigaptide: A Promising New Principle for Prevention of Arrhythmias. Basic Clin. Pharmacol. Toxicol. 2007, 101, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Shiroshita-Takeshita, A.; Sakabe, M.; Haugan, K.; Hennan, J.K.; Nattel, S. Model-dependent effects of the gap junction conduction-enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation 2007, 115, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.M.; Venkatasubramanian, S.; Vase, H.; Hyldebrandt, J.A.; Contractor, H.; Schmidt, M.R.; Bøtker, H.E.; Cruden, N.L.; Newby, D.E.; Kharbanda, R.K.; et al. Rotigaptide protects the myocardium and arterial vasculature from ischaemia reperfusion injury. Br. J. Clin. Pharmacol. 2016, 81, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl-Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Soder, B.L.; Propst, J.T.; Brooks, T.M.; Goodwin, R.L.; Friedman, H.I.; Yost, M.J.; Gourdie, R.G. The connexin43 carboxyl-terminal peptide ACT1 modulates the biological response to silicone implants. Plast. Reconstr. Surg. 2009, 123, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Figueiró, F.; de Oliveira, C.P.; Bergamin, L.S.; Rockenbach, L.; Mendes, F.B.; Jandrey, E.H.F.; Moritz, C.E.J.; Pettenuzzo, L.F.; Sévigny, J.; Guterres, S.S.; et al. Methotrexate up-regulates ecto-5′-nucleotidase/CD73 and reduces the frequency of T lymphocytes in the glioblastoma microenvironment. Purinergic Signal. 2016, 12, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.-J.; Liu, Y.; Yu, Y.-R.; Fu, Y.-Q.; Zhao, Y.; Kuang, H.-B.; Huang, Q.-R.; He, M.; Luo, D. Rutaecarpine prevented dysfunction of endothelial gap junction induced by Ox-LDL via activation of TRPV1. Eur. J. Pharmacol. 2015, 756, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fu, Y.Q.; Peng, W.J.; Yu, Y.R.; Wu, Y.S.; Yan, H.; Huang, Q.R.; He, M.; Luo, D. Rutaecarpine Reverses the Altered Connexin Expression Pattern Induced by Oxidized Low-density Lipoprotein in Monocytes. J. Cardiovasc. Pharmacol. 2016, 67, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Bikou, O.; Thomas, D.; Trappe, K.; Lugenbiel, P.; Kelemen, K.; Koch, M.; Soucek, R.; Voss, F.; Becker, R.; Katus, H.A.; et al. Connexin 43 gene therapy prevents persistent atrial fibrillation in a porcine model. Cardiovasc. Res. 2011, 92, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, H.M.; Mirabelli, P.; Xeroudaki, M.; Parekh, M.; Bertolin, M.; Breda, C.; Cagini, C.; Ponzin, D.; Lagali, N.; Ferrari, S. Effect of connexin 43 inhibition by the mimetic peptide Gap27 on corneal wound healing, inflammation and neovascularization. Br. J. Pharmacol. 2016, 173, 2880–2893. [Google Scholar] [CrossRef] [PubMed]
- Pollok, S.; Pfeiffer, A.-C.; Lobmann, R.; Wright, C.S.; Moll, I.; Martin, P.E.M.; Brandner, J.M. Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J. Cell. Mol. Med. 2011, 15, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Calder, B.W.; Rhett, J.M.; Bainbridge, H.; Fann, S.A.; Gourdie, R.G.; Yost, M.J. Inhibition of Connexin 43 Hemichannel-Mediated ATP Release Attenuates Early Inflammation During the Foreign Body Response. Tissue Eng. Part A 2015, 21, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Shiekh, G.A.; Ayub, T.; Khan, S.N.; Dar, R.; Andrabi, K.I. Reduced nitrate level in individuals with hypertension and diabetes. J. Cardiovasc. Dis. Res. 2011, 2, 172–176. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yeh, H.-I.; Lee, P.-Y.; Su, C.-H.; Tian, T.-Y.; Ko, Y.-S.; Tsai, C.-H. Reduced Expression of Endothelial Connexins 43 and 37 in Hypertensive Rats Is Rectified After 7-Day Carvedilol Treatment. Am. J. Hypertens. 2006, 19, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.B.; Mochly-Rosen, D. Nitroglycerin Use in Myocardial Infarction Patients: Risks and Benefits. Circ. J. 2012, 76, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Scheffold, N.; Herkommer, B.; Kandolf, R.; May, A.E. Lyme Carditis—Diagnosis, Treatment and Prognosis. Deutsch. Ärzteblatt Int. 2015, 112, 202–208. [Google Scholar] [CrossRef]
- Dennert, R.; Crijns, H.J.; Heymans, S. Acute viral myocarditis. Eur. Heart J. 2008, 29, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Sekhri, V.; Sanal, S.; DeLorenzo, L.J.; Aronow, W.S.; Maguire, G.P. Cardiac sarcoidosis: A comprehensive review. Arch. Med. Sci. 2011, 7, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Maury, P.; Chilon, T.; Dumonteil, N.; Fontan, A. Complete atrioventricular block persisting after regression of infectious myocarditis. J. Electrocardiol. 2008, 41, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D. Connexin’s Connection in Breast Cancer Growth and Progression. Int. J. Cell Biol. 2016, 2016, 9025905. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
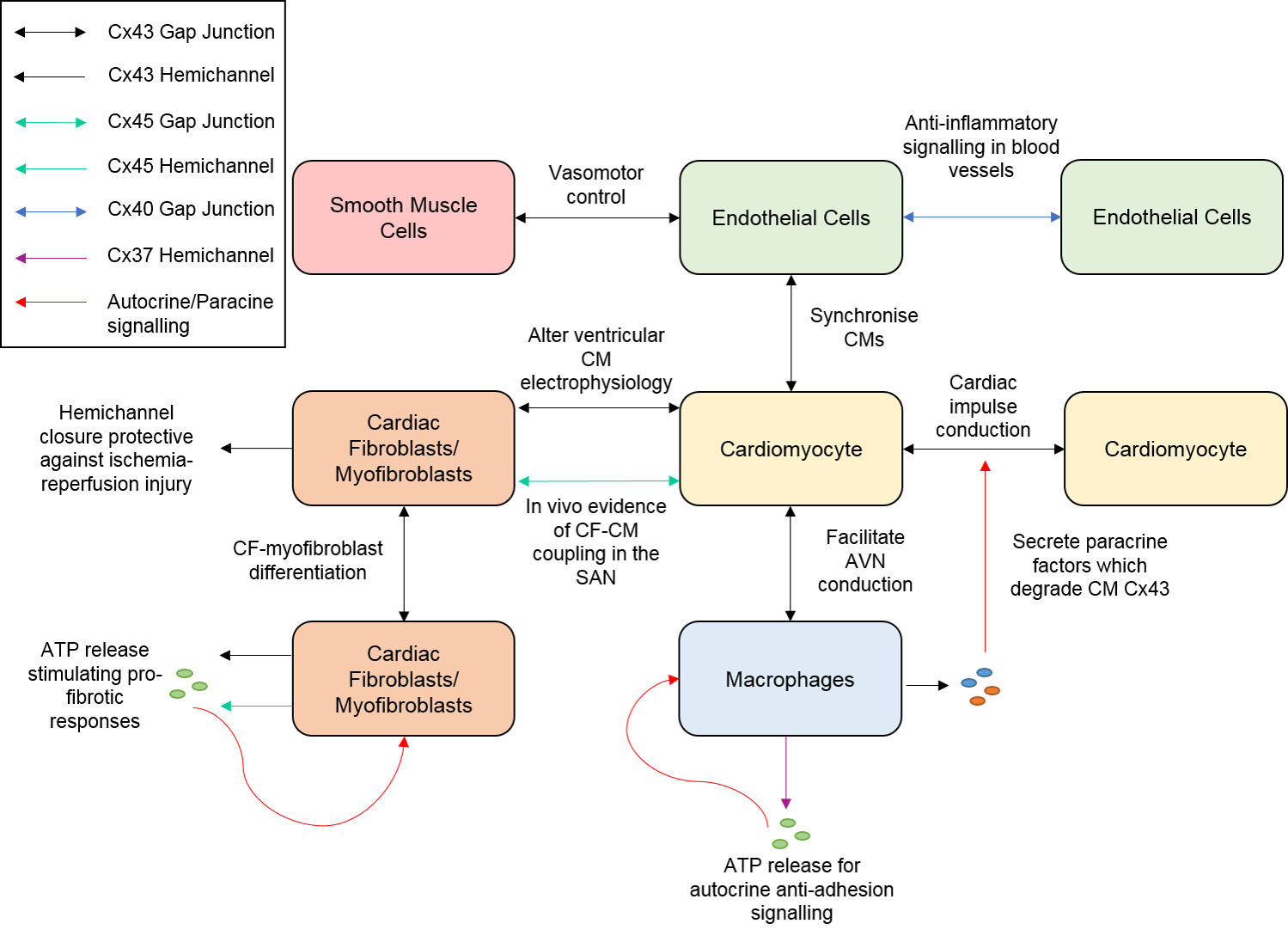{kind=link}
| Connexin Target | Therapeutic Intervention | Cell Types | Mode of Action | Potential Outcomes |
|---|---|---|---|---|
| Cx43 | Rotigaptide | Cardiomyocytes, endothelial cells | Facilitates Cx43 gap junctional coupling | Anti-arrhythmic [135,136] |
| Reduces infarct size and endothelial dysfunction following ischemia-reperfusion [137] | ||||
| Gap26 | Cardiomyocytes, myofibroblasts | Cx43 gap junction and hemichannel blocker | Reduces infarct size following ischemia-reperfusion [85] | |
| Improved CM viability following ischemia-reperfusion [84] | ||||
| Gap27 | Corneal epithelial cells, non-diabetic skin cells | Cx43 gap junction and hemichannel blocker | Improves corneal and skin wound healing [145,146] | |
| Peptide5 | Retinal pigment endothelial cells, potentially cardiac fibroblasts | Cx43 hemichannel blocker | Prevents NLRP3 inflammasome assembly and activation [88] | |
| Potential to reduce inflammatory response post-MI/HF | ||||
| Gap19 | Cardiomyocytes | Cx43 hemichannel blocker | Improves CM viability following ischemia-reperfusion [35] | |
| αCT1 | Cardiomyocytes, cardiac fibroblasts | Prevents Cx43 CT and ZO-1 interaction | Induces CF migration in vitro [81] | |
| Reduces arrhythmia following MI [138] | ||||
| Possible regenerative healing, shown in cutaneous wounds [140] | ||||
| JM2 | Endothelial cells | Cx43 hemichannel blocker | Inhibits ATP release and inflammatory response [147] | |
| Adenoviral-Cx43 gene therapy | Atrial cells | Increases expression of Cx43 | Prevents the development of persistent atrial fibrillation [144] | |
| Cx40 | Rutaecarpine | Endothelial cells | Prevents reduction in Cx40 expression | Atheroprotective [142] |
| Methotrexate (indirect effect) | Endothelial cells | CD73 activator, anti-adhesive | Methotrexate treatment shown to be atheroprotective [101] | |
| Cx37 | Rutaecarpine | Monocytes/Macrophages | Prevents reduction in Cx37 expression | Atheroprotective [143] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, R.D.; Camelliti, P. Role of Non-Myocyte Gap Junctions and Connexin Hemichannels in Cardiovascular Health and Disease: Novel Therapeutic Targets? Int. J. Mol. Sci. 2018, 19, 866. https://doi.org/10.3390/ijms19030866
Johnson RD, Camelliti P. Role of Non-Myocyte Gap Junctions and Connexin Hemichannels in Cardiovascular Health and Disease: Novel Therapeutic Targets? International Journal of Molecular Sciences. 2018; 19(3):866. https://doi.org/10.3390/ijms19030866
Chicago/Turabian StyleJohnson, Robert D., and Patrizia Camelliti. 2018. "Role of Non-Myocyte Gap Junctions and Connexin Hemichannels in Cardiovascular Health and Disease: Novel Therapeutic Targets?" International Journal of Molecular Sciences 19, no. 3: 866. https://doi.org/10.3390/ijms19030866
APA StyleJohnson, R. D., & Camelliti, P. (2018). Role of Non-Myocyte Gap Junctions and Connexin Hemichannels in Cardiovascular Health and Disease: Novel Therapeutic Targets? International Journal of Molecular Sciences, 19(3), 866. https://doi.org/10.3390/ijms19030866