Comparative Transcriptome Analyses Reveal Potential Mechanisms of Enhanced Drought Tolerance in Transgenic Salvia Miltiorrhiza Plants Expressing AtDREB1A from Arabidopsis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Illumina Sequencing and De Novo Assembly
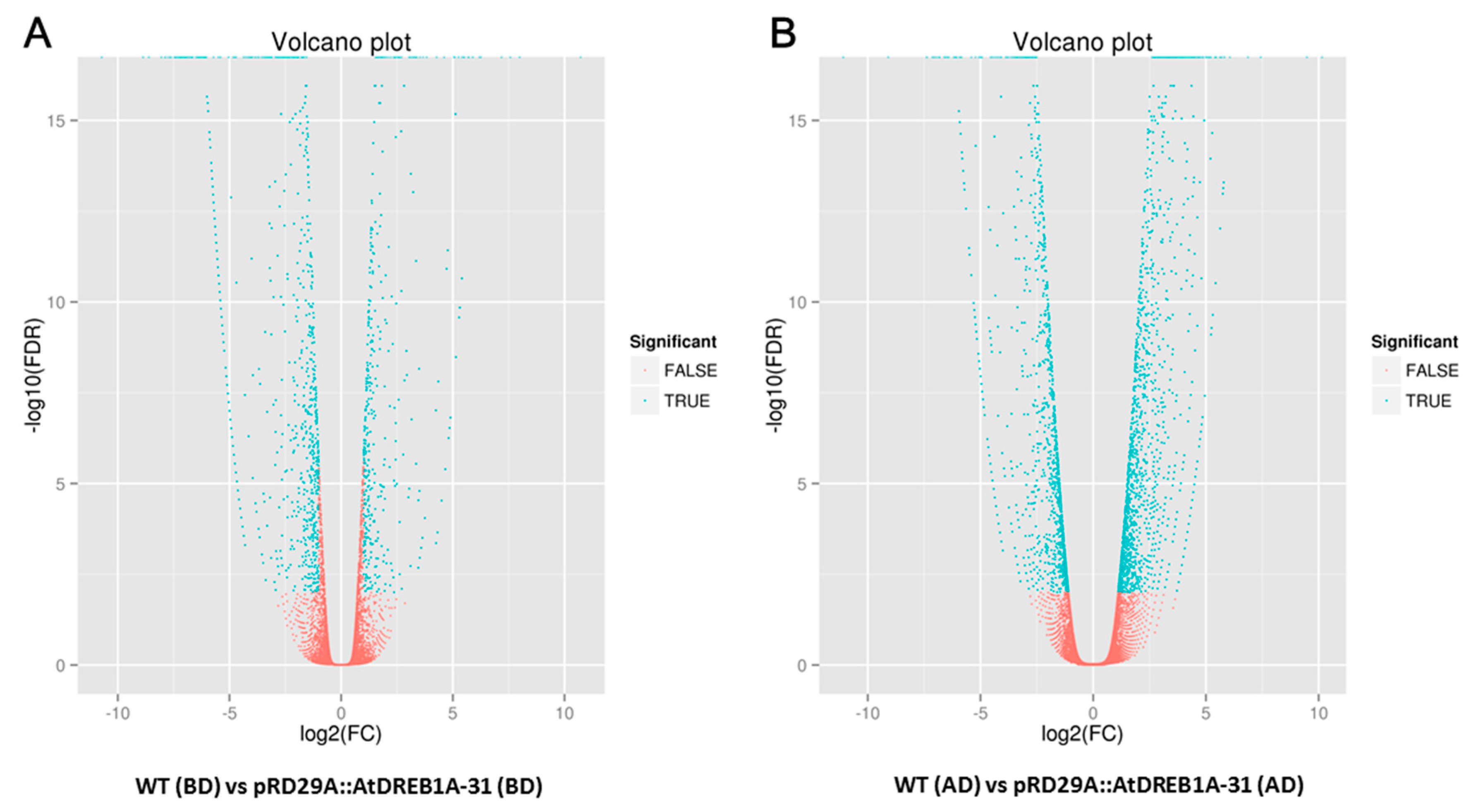
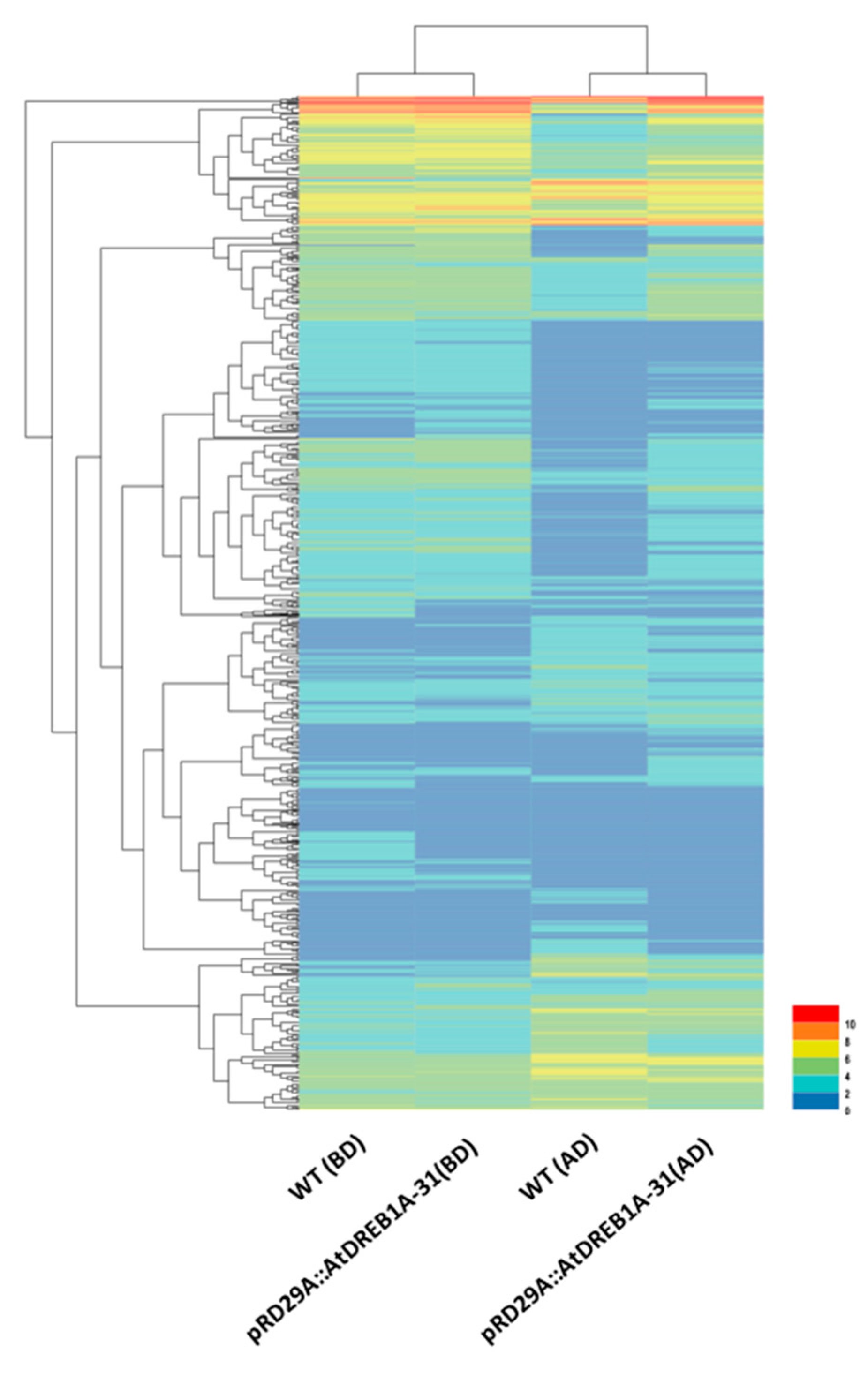
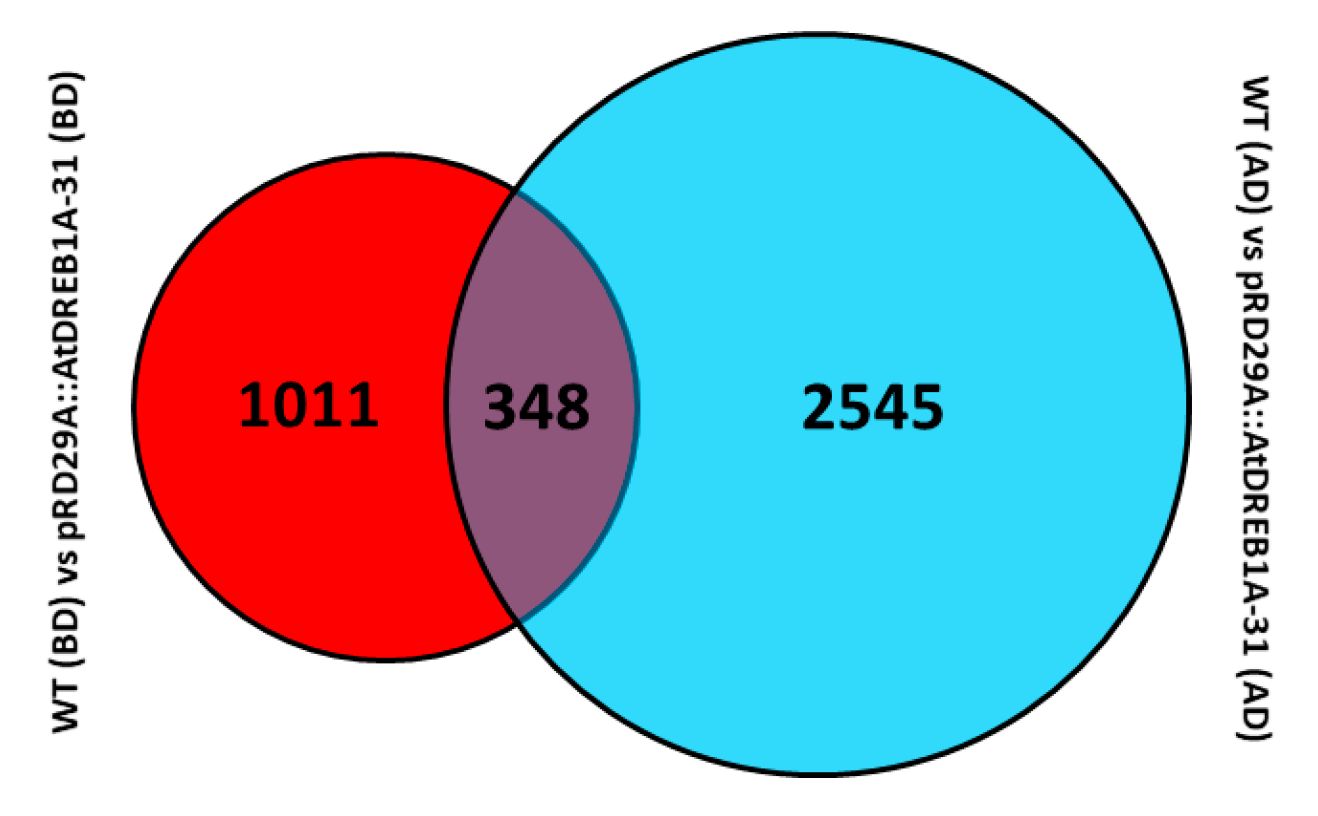
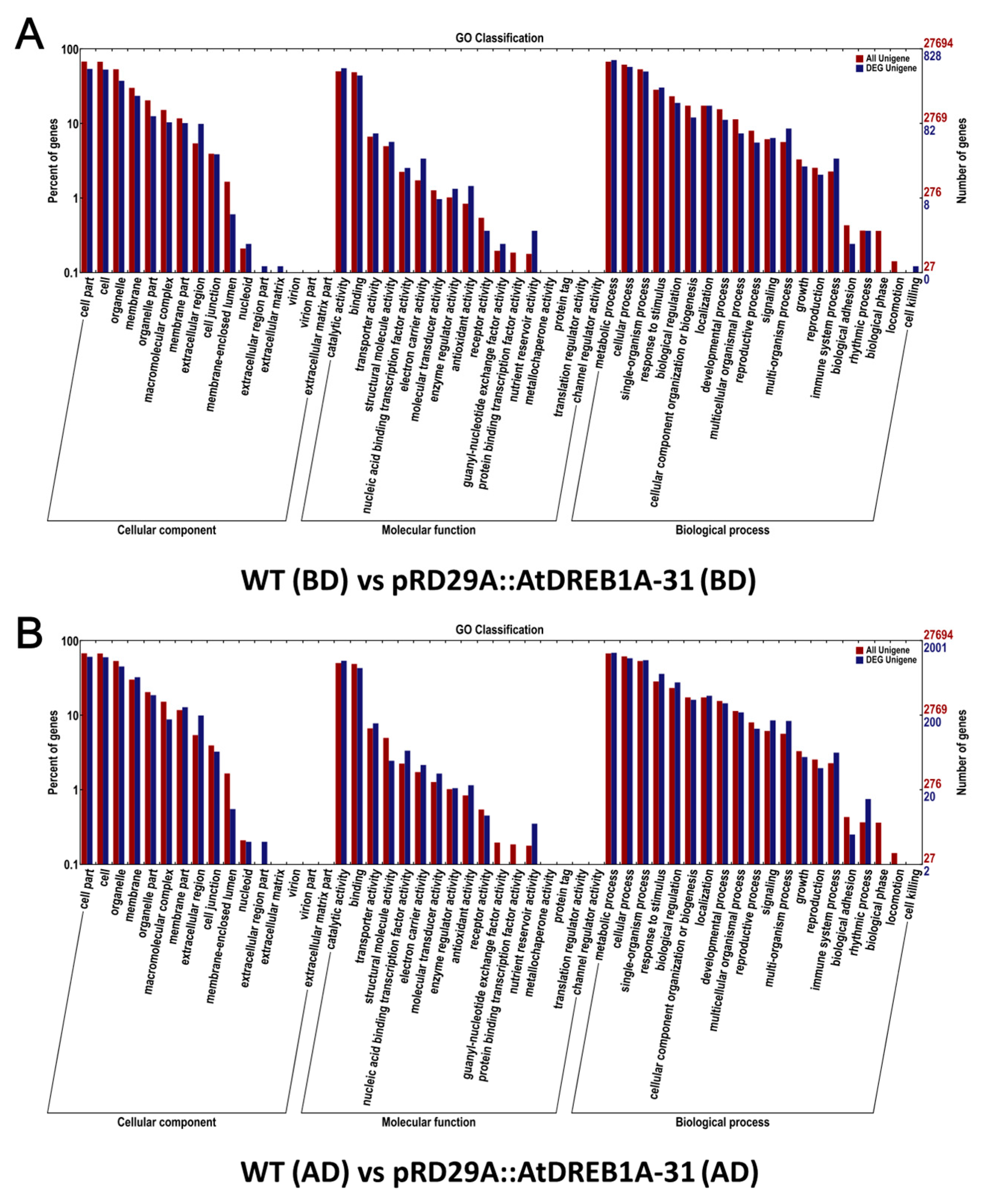
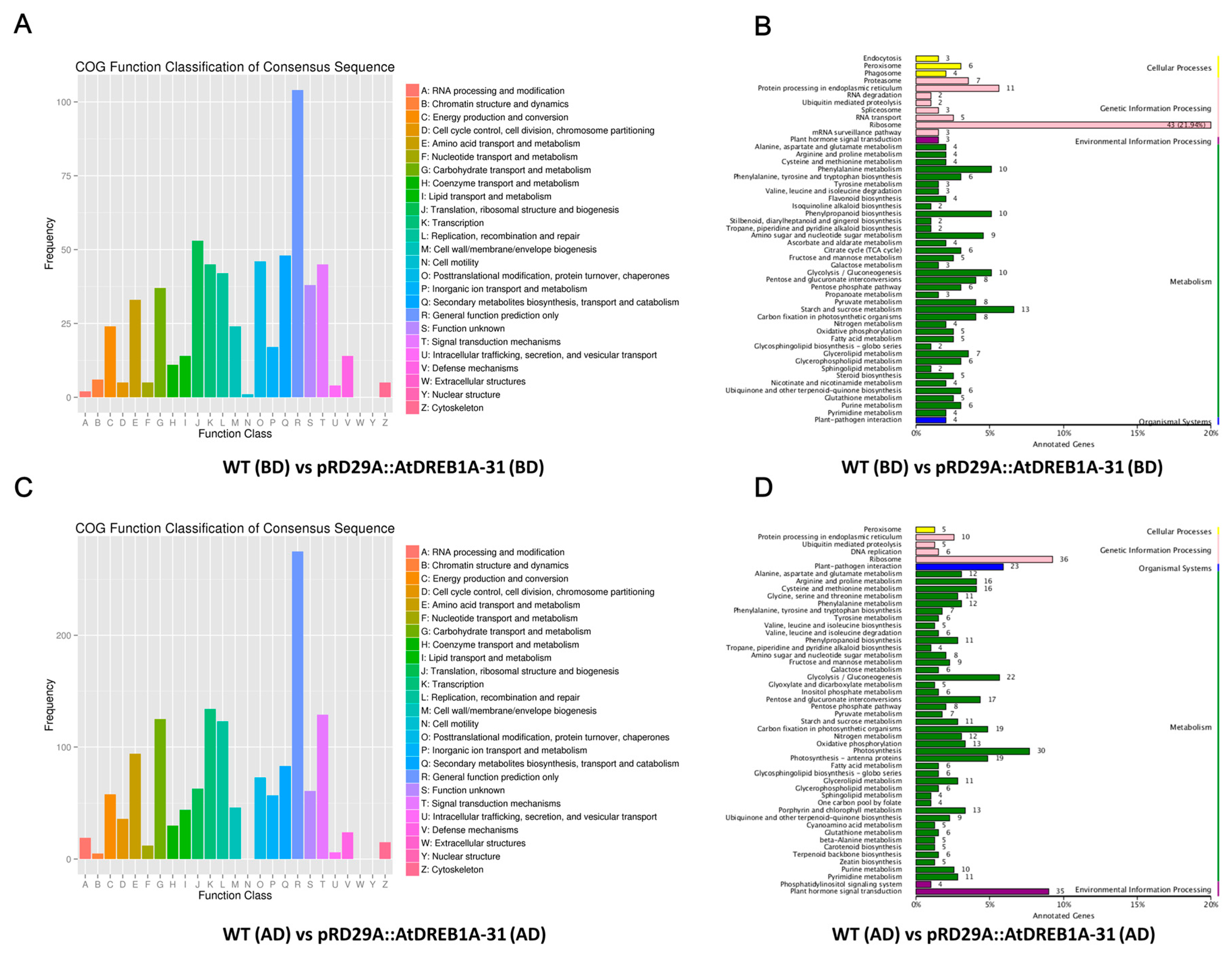
2.2. Gene Annotation and Functional Classification of the DEGs
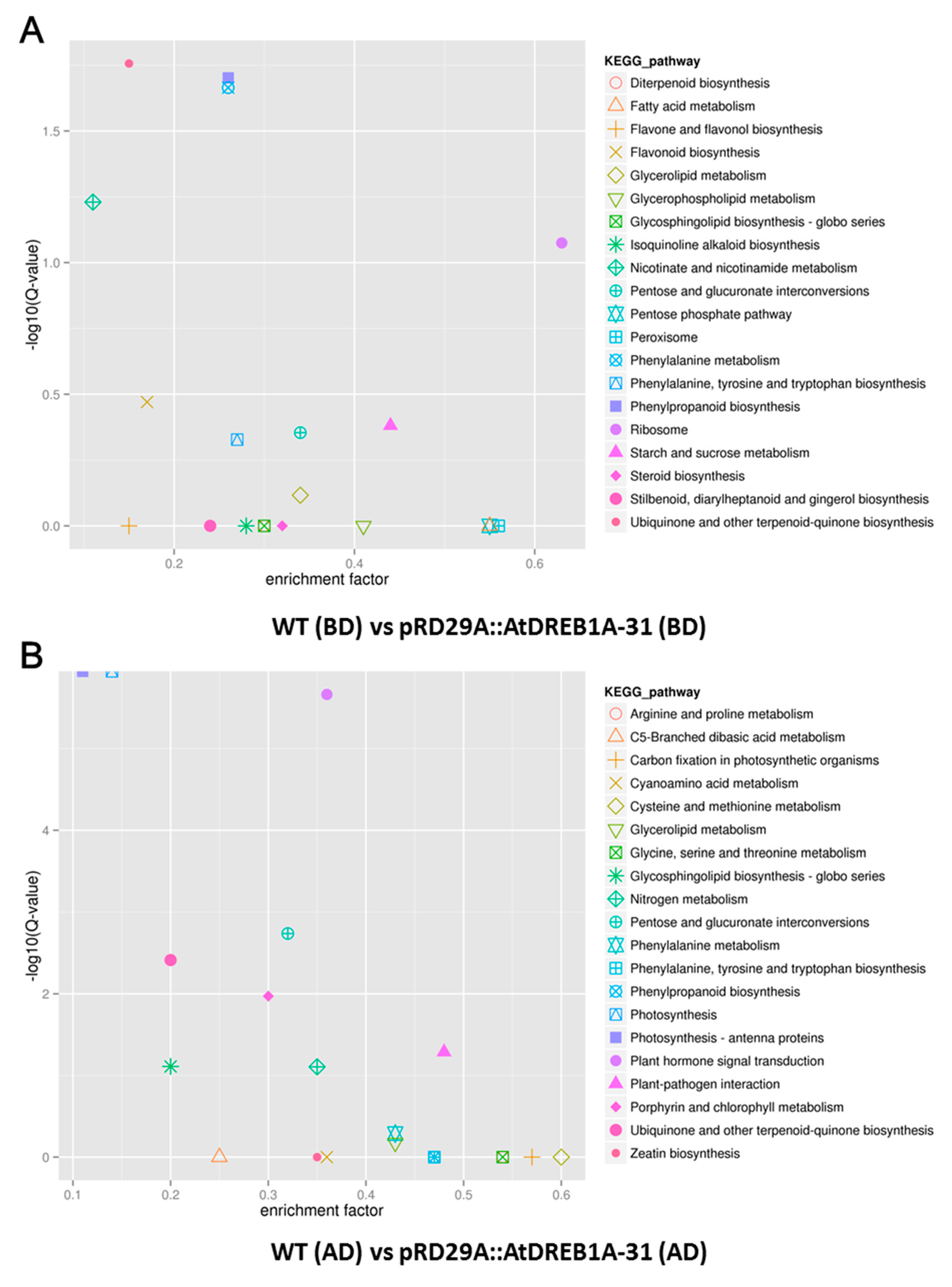
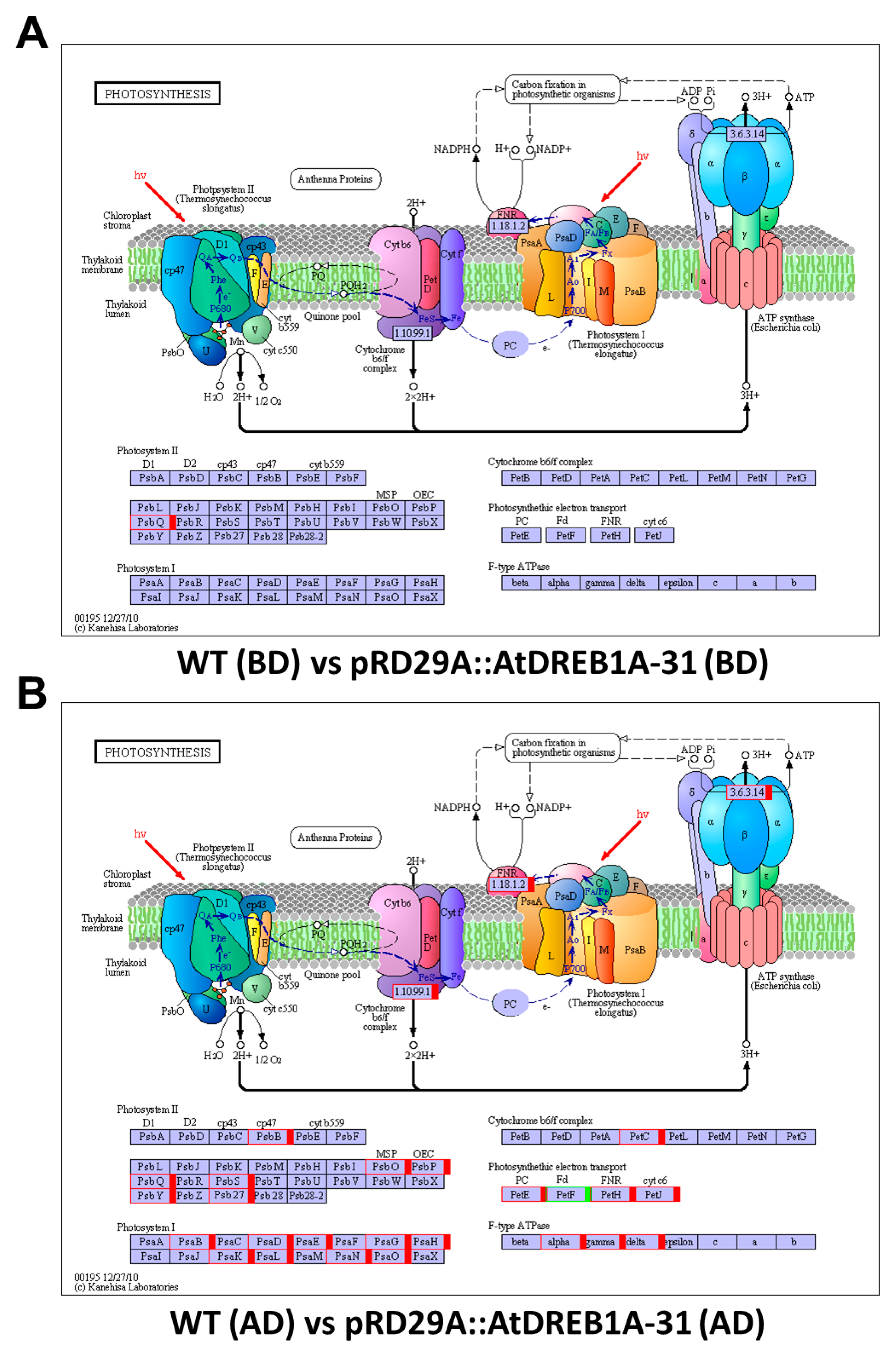
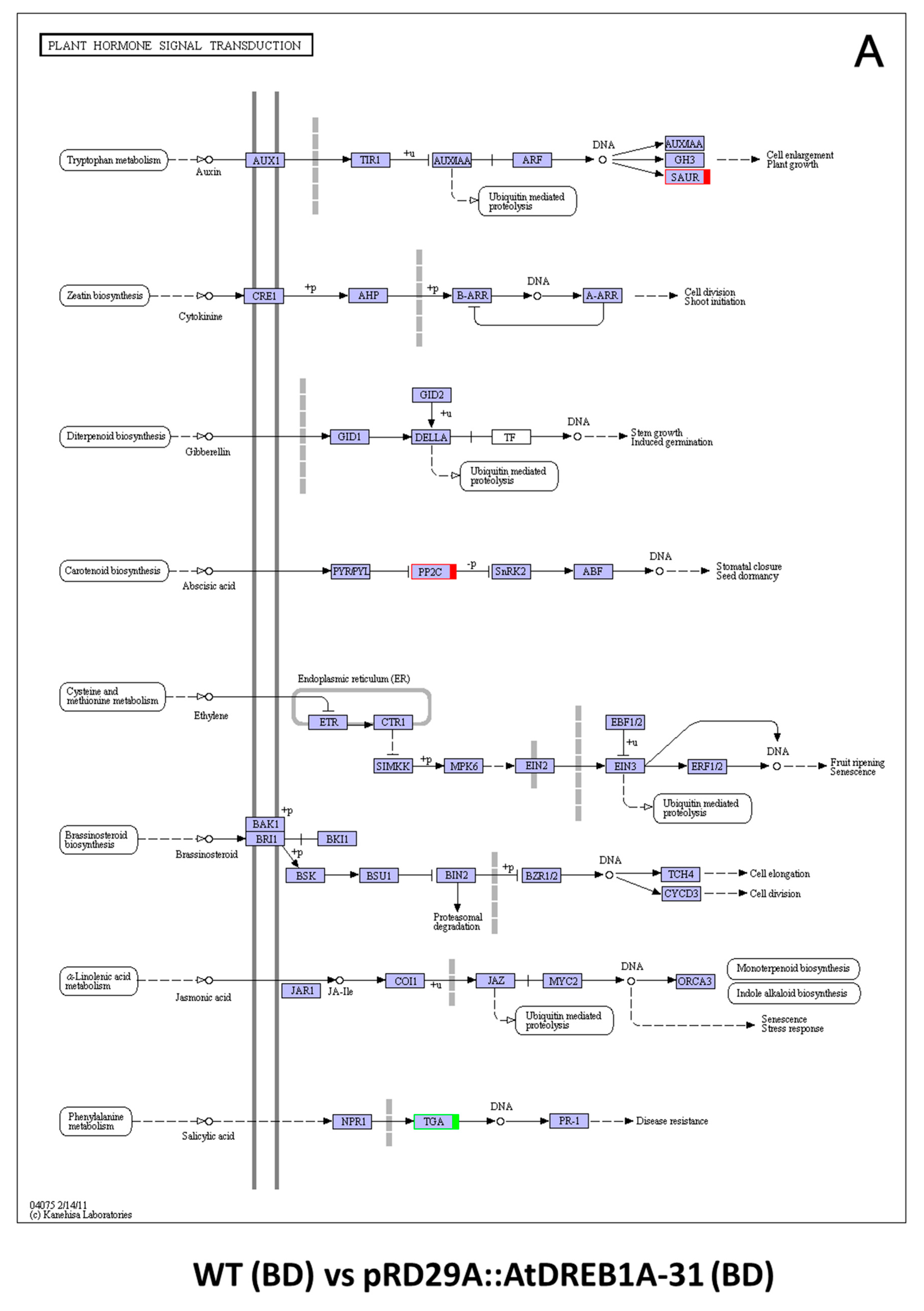
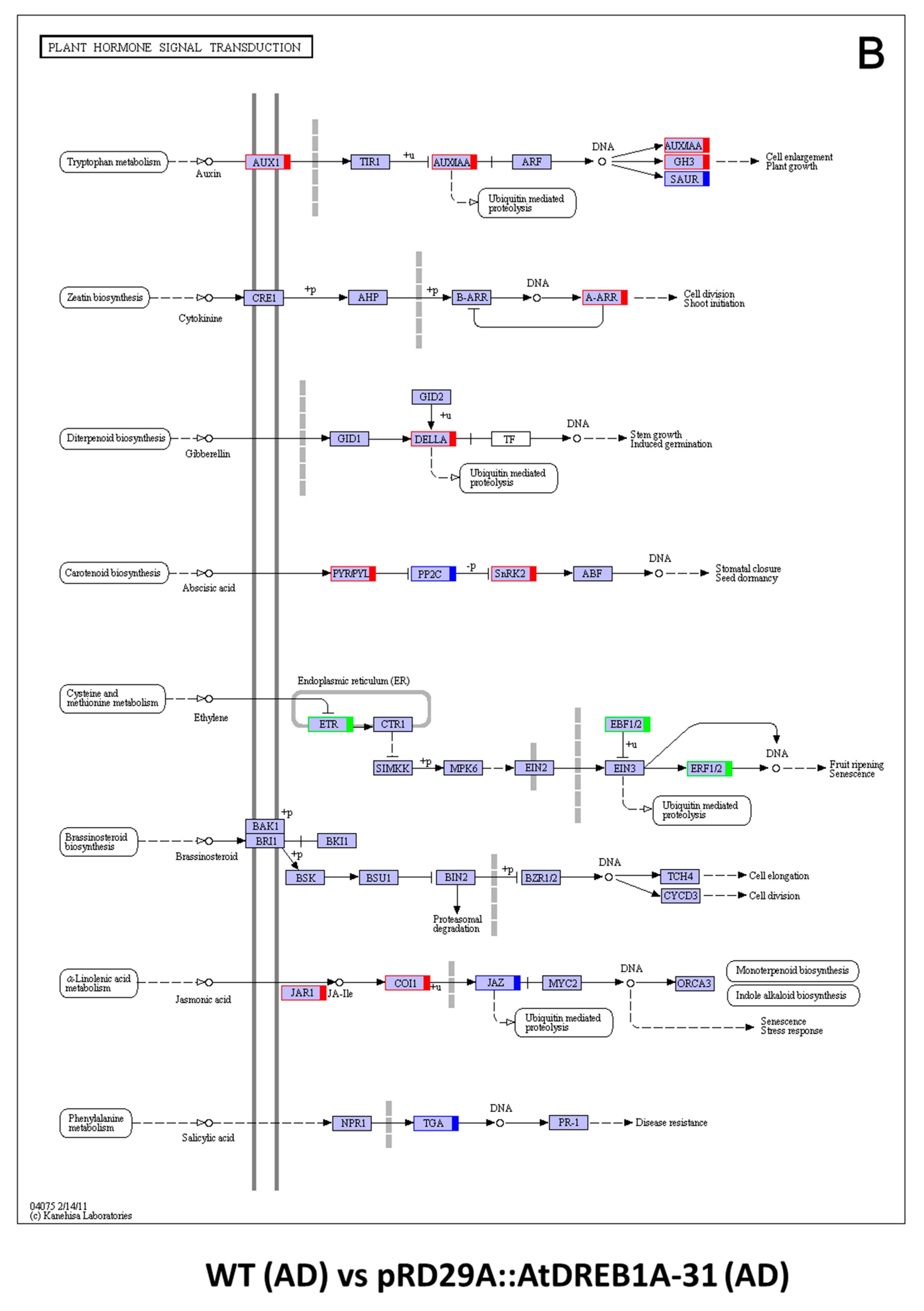
2.3. COG Enrichment and KEGG Pathway Analysis of DEGs
2.4. DEGs Related to Transcription Factors
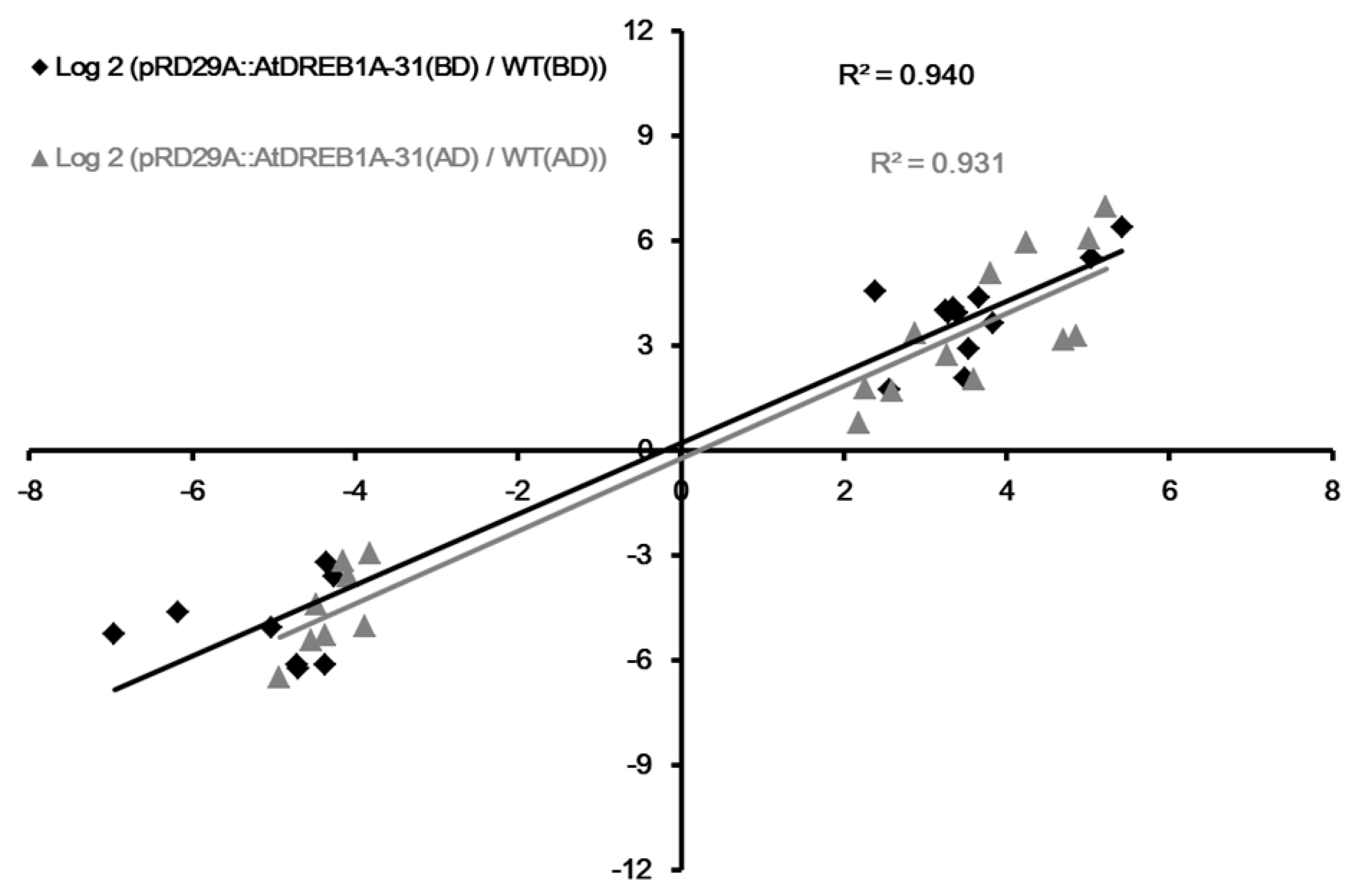
2.5. Validation of DEGs by qRT-PCR Analysis
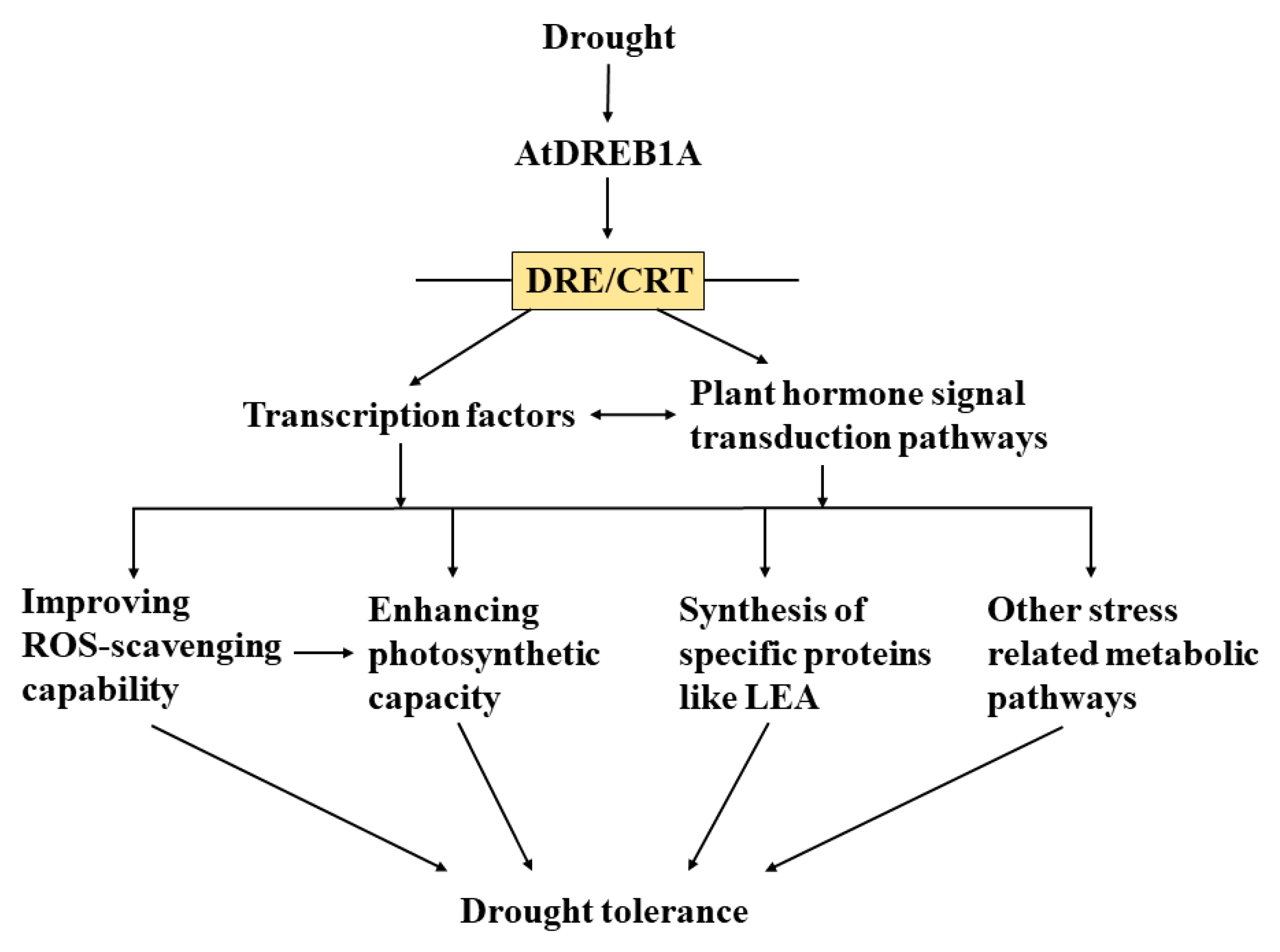
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. RNA Isolation, Library Construction and RNA Sequencing
4.3. De novo Transcriptome Assembly and Functional Annotation
4.4. Identification of DEGs
4.5. Validation of DEGs by qRT-PCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABA | abscisic acid |
| CAT | catalase |
| CK | cytokinin |
| CTAB | cetyltrimethyl ammonium bromide |
| DRE | dehydration response element |
| FC | fold change |
| JA | jasmonic acid |
| LEA | late embryogenesis abundant protein |
| MDA | malondialdehyde |
| POD | peroxidase |
| ROS | reactive oxygen species |
| RWC | relative water content |
| SA | salicylic acid |
| SOD | superoxide dismutase |
References
- Fang, J.; Lin, A.; Qiu, W.; Cai, H.; Umar, M.; Chen, R.; Ming, R. Transcriptome Profiling Revealed Stress-Induced and Disease Resistance Genes Up-Regulated in PRSV Resistant Transgenic Papaya. Front. Plant Sci. 2016, 7, 855. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Austin, R.S.; Amyot, L.; Hannoufa, A. Comparative transcriptome investigation of global gene expression changes caused by miR156 overexpression in Medicago sativa. BMC Genom. 2016, 17, 658. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.R.; Joshi, G.; Kukreja, B.; Malik, V.; Arora, P.; Pandey, R.; Shukla, R.N.; Bankar, K.G.; Katiyar-Agarwal, S.; Goel, S.; et al. Global insights into high temperature and drought stress regulated genes by RNA-Seq in economically important oilseed crop Brassica juncea. BMC Plant Biol. 2015, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Bielecka, M.; Watanabe, M.; Morcuende, R.; Scheible, W.-R.; Hawkesford, M.J.; Hesse, H.; Hoefgen, R. Transcriptome and metabolome analysis of plant sulfate starvation and resupply provides novel information on transcriptional regulation of metabolism associated with sulfur, nitrogen and phosphorus nutritional responses in Arabidopsis. Front. Plant Sci. 2014, 5, 805. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, G.; Liu, X.; Zhou, Y.; Li, Y.; Zhang, X.; Yuan, X.; Zhang, Q.; Yang, D.; Wang, T.; et al. Transcriptome Sequencing Identified Genes and Gene Ontologies Associated with Early Freezing Tolerance in Maize. Front. Plant Sci. 2016, 7, 1477. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Yan, F.; Zhang, X.; Tang, Y.; Yuan, Y. Transcriptional profiling of canola developing embryo and identification of the important roles of BnDof5.6 in embryo development and fatty acids synthesis. Plant Cell Physiol. 2015, 56, 1624–1640. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Shankar, R.; Thakkar, B.; Kudapa, H.; Krishnamurthy, L.; Mantri, N.; Varshney, R.K.; Bhatia, S.; Jain, M. Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci. Rep. 2016, 6, 19228. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, X.; Liu, J.-H. Deep sequencing-based characterization of transcriptome of trifoliate orange (Poncirus trifoliata (L.) Raf.) in response to cold stress. BMC Genom. 2015, 16, 555. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wei, W.; Pang, X.; Wang, X.; Zhang, H.; Dong, B.; Xin, Y.; Li, X.; Wang, M. Comparative transcriptome profiling of a desert evergreen shrub, Ammopiptanthus mongolicus, in response to drought and cold stresses. BMC Genom. 2014, 15, 671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Z.; Gao, J.; Wu, Y.; Xia, Z.; Zhang, H.; Wu, J. The Mechanisms of Maize Resistance to Fusarium verticillioides by Comprehensive Analysis of RNA-seq Data. Front. Plant Sci. 2016, 7, 1654. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zhu, Z.; Guo, Q.; Zhu, Y.; Yang, X.; Sun, Y. Transcriptome Analysis of Differentially Expressed Genes Provides Insight into Stolon Formation in Tulipa edulis. Front. Plant Sci. 2016, 7, 409. [Google Scholar] [CrossRef] [PubMed]
- Handa, Y.; Nishide, H.; Takeda, N.; Suzuki, Y.; Kawaguchi, M.; Saito, K. RNA-seq Transcriptional Profiling of an Arbuscular Mycorrhiza Provides Insights into Regulated and Coordinated Gene Expression in Lotus japonicus and Rhizophagus irregularis. Plant Cell Physiol. 2015, 56, 1490–1511. [Google Scholar] [CrossRef] [PubMed]
- Chan, Z.; Bigelow, P.J.; Loescher, W.; Grumet, R. Comparison of salt stress resistance genes in transgenic Arabidopsis thaliana indicates that extent of transcriptomic change may not predict secondary phenotypic or fitness effects. Plant Biotechnol. J. 2011, 10, 284–300. [Google Scholar] [CrossRef] [PubMed]
- Czemmel, S.; Höll, J.; Loyola, R.; Arce-Johnson, P.; Alcalde, J.A.; Matus, J.T.; Bogs, J. Transcriptome-Wide Identification of Novel UV-B- and Light Modulated Flavonol Pathway Genes Controlled by VviMYBF1. Front. Plant Sci. 2017, 8, 1084. [Google Scholar]
- Wang, J.; Peng, Y.; Xiao, K.; Wei, B.; Hu, J.; Wang, Z.; Song, Q.; Zhou, X. Transcriptomic response of wolf spider, Pardosa pseudoannulata, to transgenic rice expressing Bacillus thuringiensis Cry1Ab protein. BMC Biotechnol. 2017, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Kidokoro, S.; Yoshida, T.; Mizoi, J.; Todaka, D.; Fernie, A.R.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Double overexpression of DREB and PIF transcription factors improves drought stress tolerance and cell elongation in transgenic plants. Plant Biotechnol. J. 2017, 15, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Guan, C.; Liu, S.; Huang, Y.; Tian, D.; Cui, X.; Zhang, Y.; Yang, F. Comparative Transcriptomic Analyses of Differentially Expressed Genes in Transgenic Melatonin Biosynthesis Ovine HIOMT Gene in Switchgrass. Front. Plant Sci. 2016, 7, 1613. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xiu, Y.; Iqbal, A.; Zhu, C.; Wu, G.; Chang, Y.; Li, N.; Cao, Y.; Zhang, W.; Zeng, H.; Chen, S.; et al. Improvement and transcriptome analysis of root architecture by overexpression of Fraxinus pennsylvanica DREB2A transcription factor in Robinia pseudoacacia L.‘Idaho’. Plant Biotechnol. J. 2015, 14, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Lambirth, K.C.; Whaley, A.M.; Blakley, I.C.; Schlueter, J.A.; Bost, K.L.; Loraine, A.E.; Piller, K.J. A Comparison of transgenic and wild type soybean seeds: Analysis of transcriptome profiles using RNA-Seq. BMC Biotechnol. 2015, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dansana, P.K.; Kothari, K.S.; Vij, S.; Tyagi, A.K. OsiSAP1 overexpression improves water-deficit stress tolerance in transgenic rice by affecting expression of endogenous stress-related genes. Plant Cell Rep. 2014, 33, 1425–1440. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, S.; Li, H.; Jiang, J. Next-generation transcriptome analysis in transgenic birch overexpressing and suppressing APETALA1 sheds lights in reproduction development and diterpenoid biosynthesis. Plant Cell Rep. 2015, 34, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Deng, K.; Liu, D.; Gao, Y.; Liu, Y.; Yang, M.; Zhang, L.; Zheng, X.; Wang, C.; Song, W.; et al. Ectopic Expression of DREB Transcription Factor, AtDREB1A, Confers Tolerance to Drought in Transgenic Salvia miltiorrhiza. Plant Cell Physiol. 2016, 57, 1593–1609. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-J.; Song, S.I.; Kim, Y.S.; Jang, H.-J.; Kim, S.Y.; Kim, M.; Kim, Y.-K.; Nahm, B.H.; Kim, J.-K. Arabidopsis CBF3/DREB1A and ABF3 in transgenic rice increased tolerance to abiotic stress without stunting growth. Plant Physiol. 2005, 138, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M.; Miura, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. A combination of the Arabidopsis DREB1A gene and stress-inducible rd29A promoter improved drought-and low-temperature stress tolerance in tobacco by gene transfer. Plant Cell Physiol. 2004, 45, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.T.; Skinner, J.S.; Park, E.J.; Jeknić, Z.; Hayes, P.M.; Thomashow, M.F.; Chen, T.H. Use of a stress inducible promoter to drive ectopic AtCBF expression improves potato freezing tolerance while minimizing negative effects on tuber yield. Plant Biotechnol. J. 2007, 5, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Sakuma, Y.; Kasuga, M.; Ito, Y.; Seki, M.; Goda, H.; Shimada, Y.; Yoshida, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Identification of cold-inducible downstream genes of the Arabidopsis DREB1A/CBF3 transcriptional factor using two microarray systems. Plant J. 2004, 38, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Z.; Liu, J.H. Transcriptional profiling of canker-resistant transgenic sweet orange (Citrus sinensis Osbeck) constitutively overexpressing a spermidine synthase gene. Biomed. Res. Int. 2013, 2013, 918136. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Jung, I.; Shin, S.-J.; Park, J.; Rhee, S.; Kim, J.-K.; Jung, W.; Kwon, H.-B.; Kim, S. Transcriptional Network Analysis Reveals Drought Resistance Mechanisms of AP2/ERF Transgenic Rice. Front. Plant Sci. 2017, 8, 1044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Li, Y.; Yuan, Z.; He, H.; Yang, H.; Qu, H.; Ma, C.; Qu, S. Transcriptome analysis highlights defense and signaling pathways mediated by rice pi21 gene with partial resistance to Magnaporthe oryzae. Front. Plant Sci. 2016, 7, 1834. [Google Scholar] [CrossRef] [PubMed]
- Vojta, P.; Kokáš, F.; Husičková, A.; Grúz, J.; Bergougnoux, V.; Marchetti, C.F.; Jiskrová, E.; Ježilová, E.; Mik, V.; Ikeda, Y.; et al. Whole transcriptome analysis of transgenic barley with altered cytokinin homeostasis and increased tolerance to drought stress. New Biotechnol. 2016, 33, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Wakasa, Y.; Oono, Y.; Yazawa, T.; Hayashi, S.; Ozawa, K.; Handa, H.; Matsumoto, T.; Takaiwa, F. RNA sequencing-mediated transcriptome analysis of rice plants in endoplasmic reticulum stress conditions. BMC Plant Biol. 2014, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Jagadeeshselvam, N.; Valarmathi, R.; Sachin, B.; Sasikala, R.; Senthil, N.; Sudhakar, D.; Robin, S.; Muthurajan, R. Transcriptome analysis of salinity responsiveness in contrasting genotypes of finger millet (Eleusine coracana L.) through RNA-sequencing. Plant Mol. Biol. 2014, 85, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tian, Q.; Pang, T.; Jiang, L.; Wu, R.; Xia, X.; Yin, W. Deep-sequencing transcriptome analysis of low temperature perception in a desert tree, Populus euphratica. BMC Genom. 2014, 15, 326. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Niu, F.; Sun, X.; Hu, Z.; Li, X.; Ma, Y.; Zhang, H. RNA-seq analysis of unintended effects in transgenic wheat overexpressing the transcription factor GmDREB1. Crop J. 2016, 5, 207–218. [Google Scholar] [CrossRef]
- Lyamouri, M.; Enerly, E.; Lambertsson, A. Organization, sequence, and phylogenetic analysis of the ribosomal protein S3 gene from Drosophila virilis. Gene 2002, 294, 147–156. [Google Scholar] [CrossRef]
- Kang, G.Z.; Peng, H.F.; Han, Q.X.; Wang, Y.H.; Guo, T.C. Identification and expression pattern of ribosomal L5 gene in common wheat (Triticum aestivum L.). Gene 2012, 493, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Li, B.; Pandey, M.K.; Wu, Y.; Lei, Y.; Yan, L.; Dai, X.; Jiang, H.; Zhang, J.; Wei, G.; et al. Transcriptome Analysis of a New Peanut Seed Coat Mutant for the Physiological Regulatory Mechanism Involved in Seed Coat Cracking and Pigmentation. Front. Plant Sci. 2016, 7, 1491. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, L.; Hull, J.J.; Liang, S.; Daniell, H.; Jin, S.; Zhang, X. Transcriptome analysis reveals a comprehensive insect resistance response mechanism in cotton to infestation by the phloem feeding insect Bemisia tabaci (whitefly). Plant Biotechnol. J. 2016, 14, 1956–1975. [Google Scholar] [CrossRef] [PubMed]
- Cabello, J.V.; Lodeyro, A.F.; Zurbriggen, M.D. Novel perspectives for the engineering of abiotic stress tolerance in plants. Curr. Opin. Biotechnol. 2014, 26, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, H.; Wei, G.; Zhang, H.; Zhang, Y.; Gao, X. Transcriptome Profiling of hrf2 Transgenic Rapeseed Revealed Genes Related to the Defense Responses and Signaling Pathways. Crop Sci. 2015, 55, 800–810. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, Z.; Xie, S.; Si, T.; Li, Y.; Zhu, J.-K. Mutational Evidence for the Critical Role of CBF Genes in Cold Acclimation in Arabidopsis. Plant Physiol. 2016, 171, 2744. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, X.; Tang, X.M.; Xiao, L.; Sun, J.L.; Yan, X.F.; Li, D.; Deng, H.Y.; Ma, X.R. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genom. 2013, 14, 841. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Somssich, I.E. Transcriptional networks in plant immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front. Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Wolters, H.; Jürgens, G. Survival of the flexible: Hormonal growth control and adaptation in plant development. Nat. Rev. Genet. 2009, 10, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Estelle, M. Recent advances and emerging trends in plant hormone signalling. Nature 2009, 459, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol. 2011, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Wani, S.H.; Kumar, V.; Shriram, V.; Sah, S.K. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef]
- Miura, K.; Tada, Y. Regulation of water, salinity, and cold stress responses by salicylic acid. Front. Plant Sci. 2014, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, S.; Hardtke, C.S. Hormone Signalling Crosstalk in Plant Growth Regulation. Curr. Biol. 2011, 21, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.; Grewal, R.K.; Kundu, S. Regulatory Cross-Talks and Cascades in Rice Hormone Biosynthesis Pathways Contribute to Stress Signaling. Front. Plant Sci. 2016, 7, 1303. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar]
- Gutterson, N.; Reuber, T.L. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr. Opin. Plant Biol. 2004, 7, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Zhang, Z.; Chen, J.; Wang, X.; Huang, R. Expression of the ethylene response factor gene TSRF1 enhances abscisic acid responses during seedling development in tobacco. Planta 2008, 228, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Li, X.; VandenLangenberg, K.M.; Wen, D.; Sun, S.; Wei, M.; Li, Y.; Yang, F.; Shi, Q.; Wang, X. Overexpression of S-adenosyl-l-methionine synthetase increased tomato tolerance to alkali stress through polyamine metabolism. Plant Biotechnol. J. 2014, 12, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Xv, K.; Meng, Q.; Li, G.; Yang, X. Potato plants ectopically expressing Arabidopsis thaliana CBF3 exhibit enhanced tolerance to high-temperature stress. Plant Cell Environ. 2015, 38, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, J.; Liu, H.; Wang, J.; Qi, Y.; Liang, Z. Transcriptome Sequencing in Response to Salicylic Acid in Salvia miltiorrhiza. PLoS ONE 2016, 11, e0147849. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Huang, Z.; Deng, Y.; Chen, X.; Han, X.; Lai, Z. Comparative Transcriptome Analyses Reveal a Special Glucosinolate Metabolism Mechanism in Brassica alboglabra Sprouts. Front. Plant Sci. 2016, 7, 1497. [Google Scholar] [CrossRef] [PubMed]
- Laitz, A.V.N.; Acencio, M.L.; Budzinski, I.G.F.; Labate, M.T.V.; Lemke, N.; Ribolla, P.E.M.; Maia, I.G. Transcriptome Response Signatures Associated with the Overexpression of a Mitochondrial Uncoupling Protein (AtUCP1) in Tobacco. PLoS ONE 2015, 10, 6. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, T.; Deng, K.; Wang, H.; Zhang, L.; Wang, C.; Song, W.; Zhang, Y.; Chen, C. Comparative Transcriptome Analyses Reveal Potential Mechanisms of Enhanced Drought Tolerance in Transgenic Salvia Miltiorrhiza Plants Expressing AtDREB1A from Arabidopsis. Int. J. Mol. Sci. 2018, 19, 827. https://doi.org/10.3390/ijms19030827
Wei T, Deng K, Wang H, Zhang L, Wang C, Song W, Zhang Y, Chen C. Comparative Transcriptome Analyses Reveal Potential Mechanisms of Enhanced Drought Tolerance in Transgenic Salvia Miltiorrhiza Plants Expressing AtDREB1A from Arabidopsis. International Journal of Molecular Sciences. 2018; 19(3):827. https://doi.org/10.3390/ijms19030827
Chicago/Turabian StyleWei, Tao, Kejun Deng, Hongbin Wang, Lipeng Zhang, Chunguo Wang, Wenqin Song, Yong Zhang, and Chengbin Chen. 2018. "Comparative Transcriptome Analyses Reveal Potential Mechanisms of Enhanced Drought Tolerance in Transgenic Salvia Miltiorrhiza Plants Expressing AtDREB1A from Arabidopsis" International Journal of Molecular Sciences 19, no. 3: 827. https://doi.org/10.3390/ijms19030827
APA StyleWei, T., Deng, K., Wang, H., Zhang, L., Wang, C., Song, W., Zhang, Y., & Chen, C. (2018). Comparative Transcriptome Analyses Reveal Potential Mechanisms of Enhanced Drought Tolerance in Transgenic Salvia Miltiorrhiza Plants Expressing AtDREB1A from Arabidopsis. International Journal of Molecular Sciences, 19(3), 827. https://doi.org/10.3390/ijms19030827