Transcriptome Analysis of a Premature Leaf Senescence Mutant of Common Wheat (Triticum aestivum L.)



Abstract
1. Introduction
2. Results
2.1. Phenotypic Characterization and Genetic Analysis
2.2. RNA-Seq Analysis and DEGs
2.3. GO Enrichment Analysis of DEGs
2.4. KEGG Pathway Analysis of DEGs
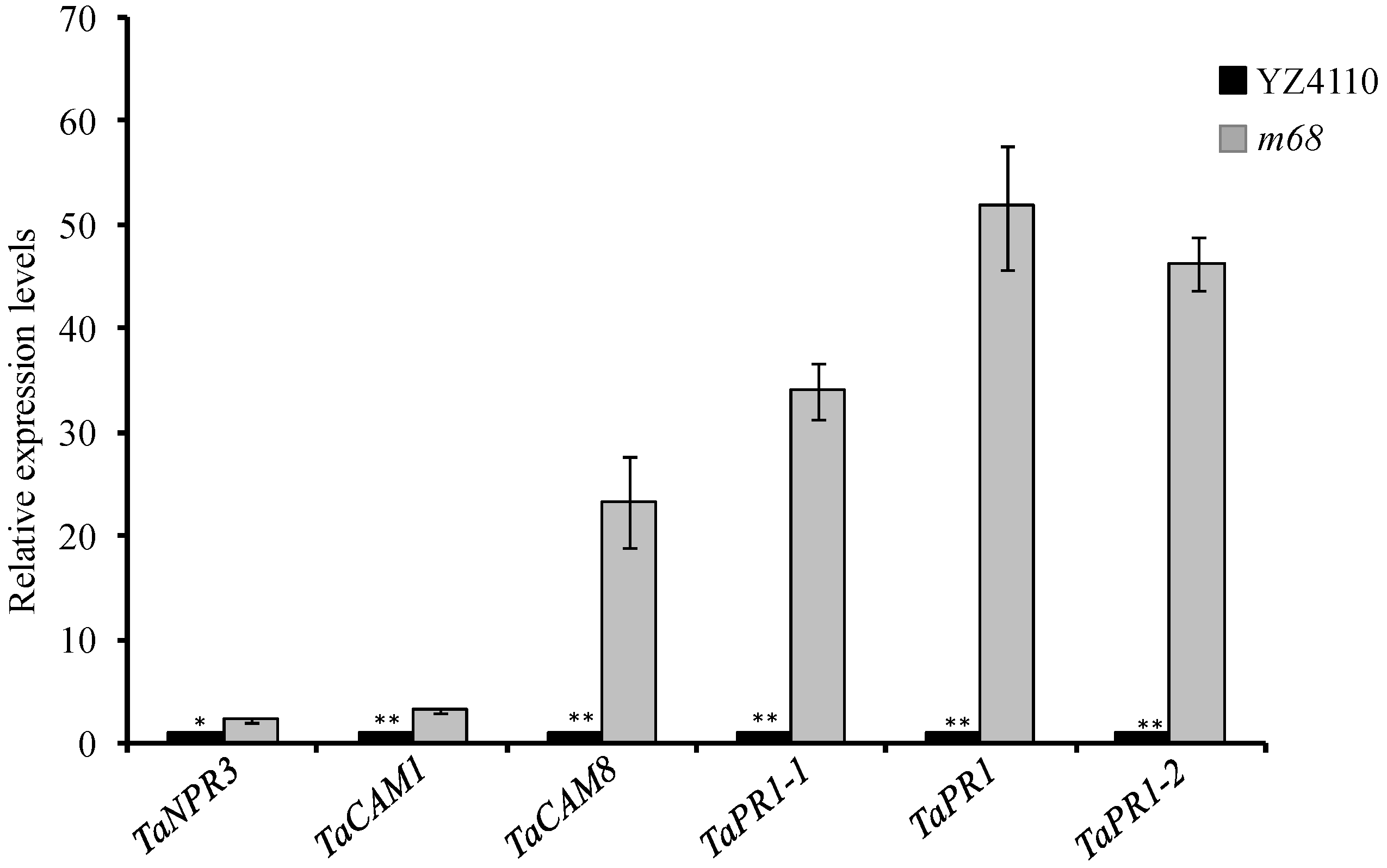
2.5. Expression Pattern Clustering Analysis of DEGs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Genetic Analysis
4.2. Plant Materials Used in RNA-Seq
4.3. Fv/Fm and MDA Content Measurement
4.4. RNA Extraction and Library Construction
4.5. Illumina Sequencing and Read Mapping
4.6. Transcriptome Analysis
4.7. Quantitative Real-Time PCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schippers, J.H.; Schmidt, R.; Wagstaff, C.; Jing, H.C. Living to die and dying to live: The survival strategy behind leaf senescence. Plant Physiol. 2015, 169, 914–930. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.L.; Culetic, A.; Boschian, L.; Krupinska, K. Plant senescence and crop productivity. Plant Mol. Biol. 2013, 82, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Distelfeld, A.; Avni, R.; Fischer, A.M. Senescence, nutrient remobilization, and yield in wheat and barley. J. Exp. Bot. 2014, 65, 3783–3798. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Woo, H.R.; Nam, H.G. Molecular genetics of leaf senescence in Arabidopsis. Trends Plant Sci. 2003, 8, 272–278. [Google Scholar] [CrossRef]
- Lira, B.S.; Gramegna, G.; Trench, B.A.; Alves, F.R.R.; Silva, E.M.; Silva, G.F.F.; Thirumalaikumar, V.P.; Lupi, A.C.D.; Demarco, D.; Purgatto, E.; et al. Manipulation of a senescence-associated gene improves fleshy fruit yield. Plant Physiol. 2017, 175, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, C. Signal transduction in leaf senescence. Plant Mol. Biol. 2013, 82, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Juvany, M.; Muller, M.; Munne-Bosch, S. Photo-oxidative stress in emerging and senescing leaves: A mirror image? J. Exp. Bot. 2013, 64, 3087–3098. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, G.; Liu, S.; An, Q.; Zheng, Q.; Li, B.; Li, Z. Comparative changes in the antioxidant system in the flag leaf of early and normally senescing near-isogenic lines of wheat (Triticum aestivum L.). Plant Cell Rep. 2014, 33, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Li, X.P.; Gan, R.; Li, P.L.; Ma, Y.Y.; Zhang, L.W.; Zhang, R.; Wang, Y.; Wang, N.N. Identification and functional characterization of a leucine-rich repeat receptor-like kinase gene that is involved in regulation of soybean leaf senescence. Plant Mol. Biol. 2006, 61, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, G.K.; Tripathy, B.C. Modulation of biosynthesis of photosynthetic pigments and light-harvesting complex in wild-type and gun5 mutant of Arabidopsis thaliana during impaired chloroplast development. Protoplasma 2016, 253, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Pudelski, B.; Soll, J.; Philippar, K. A search for factors influencing etioplast-chloroplast transition. Proc. Natl. Acad. Sci. USA 2009, 106, 12201–12206. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Balazadeh, S.; Tanaka, R.; Mueller-Roeber, B.; Tanaka, A. Overproduction of chl b retards senescence through transcriptional reprogramming in Arabidopsis. Plant Cell Physiol. 2012, 53, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.P.; Lee, T.Y.; Tanaka, A.; Charng, Y.Y. Analysis of an Arabidopsis heat-sensitive mutant reveals that chlorophyll synthase is involved in reutilization of chlorophyllide during chlorophyll turnover. Plant J. 2014, 80, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Liang, Y.; Zhang, J.; Zheng, H.; Dong, G.; Qian, Q.; Zuo, J. Involvement of a putative bipartite transit peptide in targeting rice pheophorbide a oxygenase into chloroplasts for chlorophyll degradation during leaf senescence. J. Genet. Genom. 2016, 43, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, H.; Zhai, G.; Wang, L.; Shao, J.; Tao, Y. OspTAC2 encodes a pentatricopeptide repeat protein and regulates rice chloroplast development. J. Genet. Genom. 2016, 43, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Watkins, K.P.; Miranda, R.G.; Barkan, A. The PPR-SMR protein PPR53 enhances the stability and translation of specific chloroplast RNAs in maize. Plant J. 2016, 85, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.; Guevara, D.; Yaish, M.W.; Hannam, C.; Long, N.; Clarke, J.D.; Bi, Y.M.; Rothstein, S.J. GNC and CGA1 modulate chlorophyll biosynthesis and glutamate synthase (GLU1/Fd-GOGAT) expression in Arabidopsis. PLoS ONE 2011, 6, e26765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hou, Y.; Hao, Q.; Chen, H.; Chen, L.; Yuan, S.; Shan, Z.; Zhang, X.; Yang, Z.; Qiu, D.; et al. Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS ONE 2015, 10, e0125174. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Bi, Y.M.; Rothstein, S.J. Overexpression of OsGATA12 regulates chlorophyll content, delays plant senescence and improves rice yield under high density planting. Plant Mol. Biol. 2017, 94, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.H.; Zubo, Y.O.; Tapken, W.; Kim, H.J.; Lavanway, A.M.; Howard, L.; Pilon, M.; Kieber, J.J.; Schaller, G.E. Functional characterization of the GATA transcription factors GNC and CGA1 reveals their key role in chloroplast development, growth, and division in Arabidopsis. Plant Physiol. 2012, 160, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Rozhon, W.; Poppenberger, B. The role of hormones in the aging of plants—A mini-review. Gerontology 2014, 60, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Reinbothe, C.; Springer, A.; Samol, I.; Reinbothe, S. Plant oxylipins: Role of jasmonic acid during programmed cell death, defence and leaf senescence. FEBS J. 2009, 276, 4666–4681. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.A.; Park, J.H.; Lee, G.I.; Paek, K.H.; Park, S.K.; Nam, H.G. Identification of three genetic loci controlling leaf senescence in Arabidopsis thaliana. Plant J. 1997, 12, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Kim, H.J.; Nam, H.G. Leaf senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Worley, E.; Udvardi, M. A NAP-AAO3 regulatory module promotes chlorophyll degradation via ABA biosynthesis in Arabidopsis leaves. Plant Cell 2014, 26, 4862–4874. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, Y.; Zhu, Y.; Tang, J.; Hu, B.; Liu, L.; Ou, S.; Wu, H.; Sun, X.; Chu, J.; et al. OsNAP connects abscisic acid and leaf senescence by fine-tuning abscisic acid biosynthesis and directly targeting senescence-associated genes in rice. Proc. Natl. Acad. Sci. USA 2014, 111, 10013–10018. [Google Scholar] [CrossRef] [PubMed]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC Gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Hong, S.W.; Whang, S.S.; Lim, P.O.; Nam, H.G.; Koo, J.C. Age-dependent action of an ABA-inducible receptor kinase, RPK1, as a positive regulator of senescence in Arabidopsis leaves. Plant Cell Physiol. 2011, 52, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.N.; Lee, K.H.; Kim, C.S. Abscisic acid receptor pyrabactin resistance-like 8, PYL8, is involved in glucose response and dark-induced leaf senescence in Arabidopsis. Biochem. Biophys. Res. Commun. 2015, 463, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Besseau, S.; Li, J.; Palva, E.T. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Park, H.C.; Park, C.Y.; Koo, S.C.; Cheong, M.S.; Kim, K.E.; Kim, M.C.; Lim, C.O.; Lee, S.Y.; Yun, D.J.; Chung, W.S. AtCML8, a calmodulin-like protein, differentially activating CaM-dependent enzymes in Arabidopsis thaliana. Plant Cell Rep. 2010, 29, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ryu, H.; Hong, S.H.; Woo, H.R.; Lim, P.O.; Lee, I.C.; Sheen, J.; Nam, H.G.; Hwang, I. Cytokinin-mediated control of leaf longevity by AHK3 through phosphorylation of ARR2 in Arabidopsis. Proc. Natl. Acad. Sci. USA 2006, 103, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, W.; Gan, S.S. SAUR36, a small auxin up RNA gene, is involved in the promotion of leaf senescence in Arabidopsis. Plant Physiol. 2013, 161, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.M.; Zhu, T.; Rothstein, S.J. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 2009, 151, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chan, Z.; Gao, J.; Xing, L.; Cao, M.; Yu, C.; Hu, Y.; You, J.; Shi, H.; Zhu, Y.; et al. ABA receptor PYL9 promotes drought resistance and leaf senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Liu, L.L.; Wang, C.; Gan, T.; Zhang, Z.; Wang, Y.; Wang, D.; Niu, M.; Long, W.; et al. Isolation and characterization of a spotted leaf 32 mutant with early leaf senescence and enhanced defense response in rice. Sci. Rep. 2017, 7, 41846. [Google Scholar] [CrossRef] [PubMed]
- Krattinger, S.G.; Lagudah, E.S.; Spielmeyer, W.; Singh, R.P.; Huerta-Espino, J.; McFadden, H.; Bossolini, E.; Selter, L.L.; Keller, B. A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 2009, 323, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Checovich, M.L.; Galatro, A.; Moriconi, J.I.; Simontacchi, M.; Dubcovsky, J.; Santa-Maria, G.E. The stay-green phenotype of TaNAM-RNAi wheat plants is associated with maintenance of chloroplast structure and high enzymatic antioxidant activity. Plant Physiol. Biochem. 2016, 104, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, S.; Xia, G. From genome to gene: A new epoch for wheat research? Trends Plant Sci. 2015, 20, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.K.; Lahiri, A. Specificity determinants for the abscisic acid response element. FEBS Open Bio 2013, 3, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Sugiyama, N.; Mizoguchi, M.; Hayashi, S.; Myouga, F.; Yamaguchi-Shinozaki, K.; Ishihama, Y.; Hirayama, T.; Shinozaki, K. Type 2C protein phosphatases directly regulate abscisic acid-activated protein kinases in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 17588–17593. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiang, S.; Chen, Y.; Li, D.; Yu, D. Arabidopsis WRKY45 interacts with the DELLA protein RGL1 to positively regulate age-triggered leaf senescence. Mol. Plant 2017, 10, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Song, W.M.; Pan, J.; Jiang, C.M.; Srivastava, R.; Li, B.; Zhu, L.Y.; Su, H.Y.; Gao, X.S.; Liu, H.; et al. Overexpression of the NDR1/HIN1-like gene NHL6 modifies seed germination in response to abscisic acid and abiotic atresses in Arabidopsis. PLoS ONE 2016, 11, e0148572. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Jaradat, M.R.; Wu, W.; Ambrose, S.J.; Ross, A.R.; Abrams, S.R.; Cutler, A.J. Structural analogs of ABA reveal novel features of ABA perception and signaling in Arabidopsis. Plant J. 2007, 50, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Dreher, K.A.; Brown, J.; Saw, R.E.; Callis, J. The Arabidopsis Aux/IAA protein family has diversified in degradation and auxin responsiveness. Plant Cell 2006, 18, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, L.; Na, X.; You, J.; Hu, W.; Liang, X.; Liu, J.; Mao, L.; Wang, X.; Wang, H.; et al. Narciclasine inhibits the responses of Arabidopsis roots to auxin. Planta 2012, 236, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Ohto, M.A.; Hayashi, S.; Sawa, S.; Hashimoto-Ohta, A.; Nakamura, K. Involvement of HLS1 in sugar and auxin signaling in Arabidopsis leaves. Plant Cell Physiol. 2006, 47, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- McGarry, R.C.; Ayre, B.G. A DNA element between At4g28630 and At4g28640 confers companion-cell specific expression following the sink-to-source transition in mature minor vein phloem. Planta 2008, 228, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.E.; Li, J.; Argueso, C.; Gonzalez, M.; Lee, E.; Lewis, M.W.; Maxwell, B.B.; Perdue, T.D.; Schaller, G.E.; Alonso, J.M.; et al. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell 2006, 18, 3073–3087. [Google Scholar] [CrossRef] [PubMed]
- Cortleven, A.; Marg, I.; Yamburenko, M.V. Cytokinin regulates the etioplast-chloroplast transition through the two-component signaling system and activation of chloroplast-related genes. Plant Physiol. 2016, 172, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yoo, M.J.; Koh, J.; Liu, L.; Chen, Y.; Acikgoz, D.; Wang, Q.; Chen, S. Molecular reprogramming of Arabidopsis in response to perturbation of jasmonate signaling. J. Proteome Res. 2014, 13, 5751–5766. [Google Scholar] [CrossRef] [PubMed]
- Danon, A.; Miersch, O.; Felix, G.; Camp, R.G.; Apel, K. Concurrent activation of cell death-regulating signaling pathways by singlet oxygen in Arabidopsis thaliana. Plant J. 2005, 41, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Q.; Yan, S.; Saleh, A.; Wang, W.; Ruble, J.; Oka, N.; Mohan, R.; Spoel, S.H.; Tada, Y.; Zheng, N.; et al. NPR3 and NPR4 are receptors for the immune signal salicylic acid in plants. Nature 2012, 486, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Lee, D.H.; Hwang, B.K. The pepper calmodulin gene CaCaM1 is involved in reactive oxygen species and nitric oxide generation required for cell death and the defense response. Mol. Plant Microbe Interact. 2009, 22, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Robe, E.; Jomat, L.; Aldon, D.; Mazars, C.; Galaud, J.P. CML8, an Arabidopsis calmodulin-like protein, plays a role in pseudomonas syringae plant immunity. Plant Cell Physiol. 2017, 58, 307–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schon, M.; Toller, A.; Diezel, C.; Roth, C.; Westphal, L.; Wiermer, M.; Somssich, I.E. Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4. Mol. Plant Microbe Interact. 2013, 26, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Truman, W.; de Zabala, M.T.; Grant, M. Type III effectors orchestrate a complex interplay between transcriptional networks to modify basal defence responses during pathogenesis and resistance. Plant J. 2006, 46, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S. Molecular regulation of leaf senescence. Curr. Opin. Plant Biol. 2003, 6, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hao, Q.; Tian, F.; Li, Q.; Wang, W. The stay-green phenotype of wheat mutant tasg1 is associated with altered cytokinin metabolism. Plant Cell Rep. 2016, 35, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Saher, S.; Piqueras, A.; Hellin, E.; Olmos, E. Hyperhydricity in micropropagated carnation shoots: The role of oxidative stress. Physiol. Plant 2004, 120, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Years | Location | No. # | Plant Height | Spike Length | Effective Tiller Number | Spikelet Number | Grain Number per Spike | Floret Number | 1000-Grain Weight (g) |
|---|---|---|---|---|---|---|---|---|---|---|
| YZ4110 | 2014 | Beijing | 30 | 68.9 ± 1.1 | 9.9 ± 0.1 | 13.2 ± 0.2 | 24.9 ± 0.1 | 60.6 ± 4.5 | 79.5 ± 11.5 | 49.4 ± 0.8 |
| m68 | 30 | 61.4 ± 0.5 ** | 8.8 ± 0.9 | 7.3 ± 1.3 ** | 17.3 ± 0.8 ** | 27.8 ± 3.9 ** | 50.8 ± 6.8 ** | 15.3 ± 0.5 ** | ||
| YZ4110 | 2015 | Beijing | 30 | 66.8 ± 0.4 | 9.1 ± 0.8 | 15.3 ± 4.9 | 22.8 ± 0.7 | 48.7 ± 4.1 | 69.2 ± 7.8 | 50.9 ± 0.9 |
| m68 | 30 | 57.4 ± 0.7 ** | 8.9 ± 0.1 | 8.1 ± 0.7 ** | 20.2 ± 0.1 * | 33.9 ± 0.1 ** | 59.7 ± 8.4 * | 15.7 ± 0.5 ** | ||
| YZ4110 | 2016 | Beijing | 30 | 70.5 ± 1.7 | 8.9 ± 0.4 | 16.7 ± 2.4 | 23.1 ± 0.4 | 49.8 ± 2.9 | 72.8 ± 3.8 | 48.0 ± 0.9 |
| m68 | 30 | 57.8 ± 1.5 ** | 9.0 ± 0.4 | 6.2 ± 0.5 ** | 19.9 ± 0.5 * | 37.3 ± 3.1 ** | 63.6 ± 8.5 * | 15.7 ± 0.8 ** |
| Sample ID | No. of Total Reads (Mb) | Read Length (bp) | Data Size (G) | Q30 (%) | GC (%) | Mappable Reads (%) | Unique Mapped Reads (%) | Multiple Mapped Reads (%) |
|---|---|---|---|---|---|---|---|---|
| WT_S1 | 41.3 | 150 | 6.19 | 95.71 | 55.66 | 88.92 | 81.33 | 7.59 |
| WT_S2 | 40.8 | 150 | 6.12 | 94.93 | 56.33 | 89.69 | 78.56 | 8.40 |
| WT_S3 | 40.9 | 150 | 6.13 | 95.31 | 55.00 | 89.34 | 79.57 | 9.77 |
| WT_S4 | 41.5 | 150 | 6.23 | 95.48 | 55.00 | 87.88 | 79.59 | 8.29 |
| M_S1 | 41.4 | 150 | 6.21 | 94.65 | 56.00 | 85.72 | 77.81 | 7.91 |
| M_S2 | 40.6 | 150 | 6.13 | 94.84 | 55.33 | 85.61 | 79.64 | 5.97 |
| M_S3 | 41.5 | 150 | 6.22 | 95.85 | 54.33 | 87.89 | 81.69 | 6.20 |
| M_S4 | 41.3 | 150 | 6.19 | 95.69 | 53.66 | 88.75 | 82.53 | 6.22 |
| Wheat Gene ID | Gene Name | E-Value | LogFC | Participate in Signal Transduction Pathway | Reference |
|---|---|---|---|---|---|
| MSTRG.35870.1 | ABF1 | 4.00 × 10−35 | −2.08 | ABA signaling | [40] |
| MSTRG.10319.1 | OST1 | 1.00 × 10−142 | −2.19 | ABA signaling | [35] |
| MSTRG.11222.1 | SNF1-like | 1.00 × 10−142 | −2.11 | ABA signaling | [41] |
| MSTRG.14188.1 | HAB1 | 6.00 × 10−63 | −3.61 | ABA signaling | [42] |
| MSTRG.33951.1 | ABF4 | 1.00 × 10−35 | −2.66 | ABA signaling | [43] |
| MSTRG.18662.1 | HAI3 | 8.00 × 10−63 | −4.07 | ABA signaling | [44] |
| MSTRG.36133.1 | IAA13 | 3.00 × 10−39 | 2.31 | AUX signaling | [45] |
| MSTRG.30418.1 | IAA7 | 4.00 × 10−47 | 2.89 | AUX signaling | [46] |
| MSTRG.11304.1 | SAAUR3 | 0 | −2.53 | AUX signaling | [47] |
| MSTRG.37877.1 | IAA11 | 3.00 × 10−15 | 2.12 | AUX signaling | [48] |
| MSTRG.10089.1 | SAUR38 | 2.00 × 10−22 | −5.79 | AUX signaling | — |
| MSTRG.7713.1 | SAUR38 | 4.00 × 10−20 | −9.86 | AUX signaling | — |
| MSTRG.41779.1 | SAUR71 | 4.00 × 10−13 | −3.23 | AUX signaling | — |
| MSTRG.3553.1 | AHP4 | 1.00 × 10−45 | 3.37 | CK signaling | [49] |
| MSTRG.40221.1 | ARR12 | 2.00 × 10−93 | 2.68 | CK signaling | [50] |
| MSTRG.21216.1 | EIN2 | 1.00 × 10−180 | 2.07 | Ethylene signal | [18] |
| MSTRG.31221.1 | JAZ2 | 4.00 × 10−12 | −3.69 | JA signaling | [51] |
| MSTRG.38578.1 | PR1-like | 1.00 × 10−39 | −3.94 | JA signaling | [52] |
| MSTRG.18976.1 | NPR3 | 1.00 × 10−106 | −1.23 | SA signaling | [53] |
| MSTRG.13794.1 | CAM1 | 2.00 × 10−81 | −2.20 | SA signaling | [54] |
| MSTRG.27143.1 | CAM8 | 2.00 × 10−54 | −4.39 | SA signaling | [55] |
| MSTRG.29737.1 | PR1-like | 7.00 × 10−39 | −8.88 | SA signaling | — |
| MSTRG.32097.1 | PR1-like | 7.00 × 10−37 | −7.75 | SA signaling | — |
| MSTRG.26416.1 | PR1-like | 4.00 × 10−39 | −5.92 | SA signaling | — |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Xia, C.; Zhang, L.; Dong, C.; Liu, X.; Kong, X. Transcriptome Analysis of a Premature Leaf Senescence Mutant of Common Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2018, 19, 782. https://doi.org/10.3390/ijms19030782
Zhang Q, Xia C, Zhang L, Dong C, Liu X, Kong X. Transcriptome Analysis of a Premature Leaf Senescence Mutant of Common Wheat (Triticum aestivum L.). International Journal of Molecular Sciences. 2018; 19(3):782. https://doi.org/10.3390/ijms19030782
Chicago/Turabian StyleZhang, Qiang, Chuan Xia, Lichao Zhang, Chunhao Dong, Xu Liu, and Xiuying Kong. 2018. "Transcriptome Analysis of a Premature Leaf Senescence Mutant of Common Wheat (Triticum aestivum L.)" International Journal of Molecular Sciences 19, no. 3: 782. https://doi.org/10.3390/ijms19030782
APA StyleZhang, Q., Xia, C., Zhang, L., Dong, C., Liu, X., & Kong, X. (2018). Transcriptome Analysis of a Premature Leaf Senescence Mutant of Common Wheat (Triticum aestivum L.). International Journal of Molecular Sciences, 19(3), 782. https://doi.org/10.3390/ijms19030782