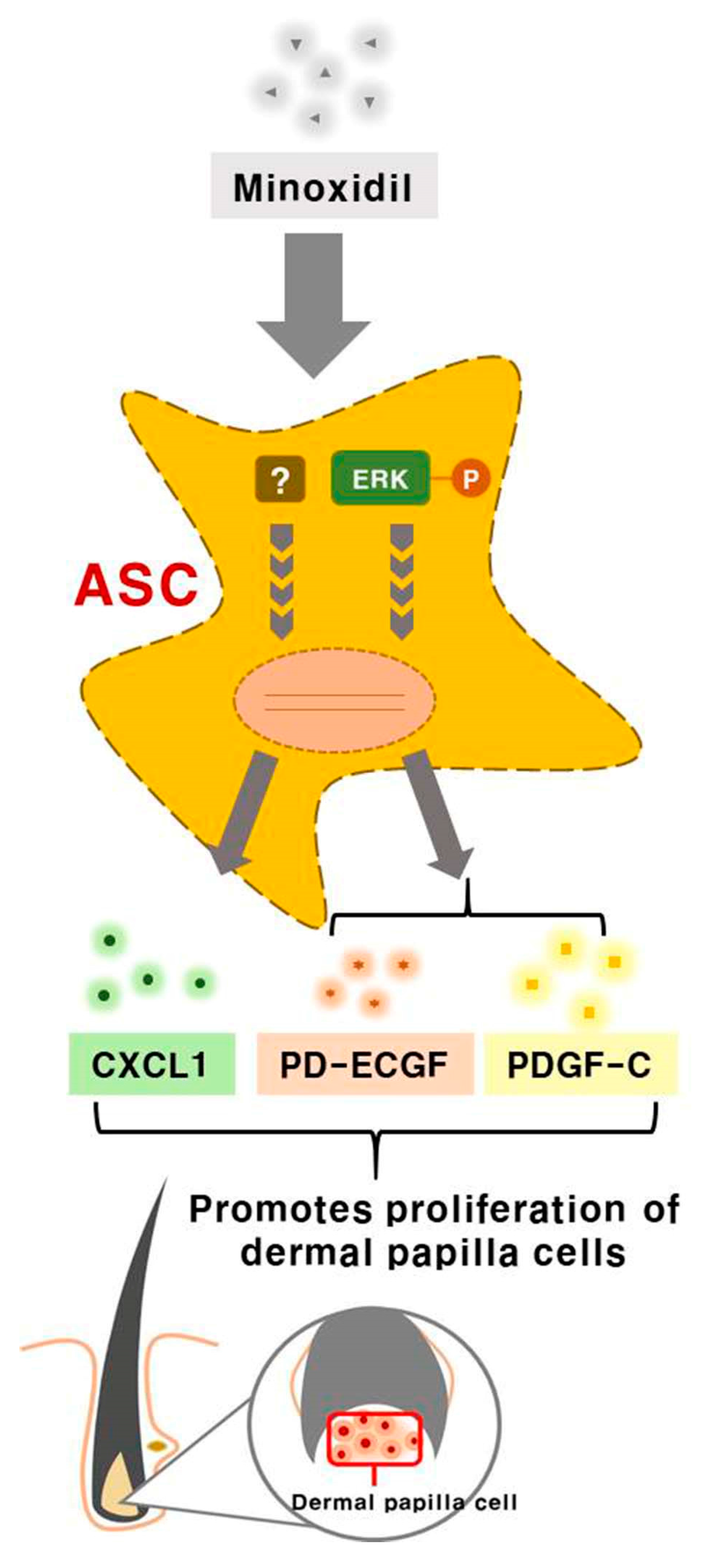
Minoxidil Promotes Hair Growth through Stimulation of Growth Factor Release from Adipose-Derived Stem Cells



Abstract
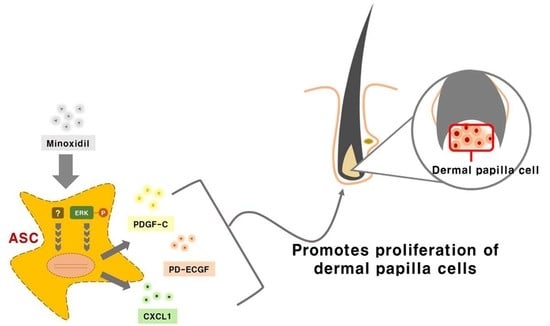
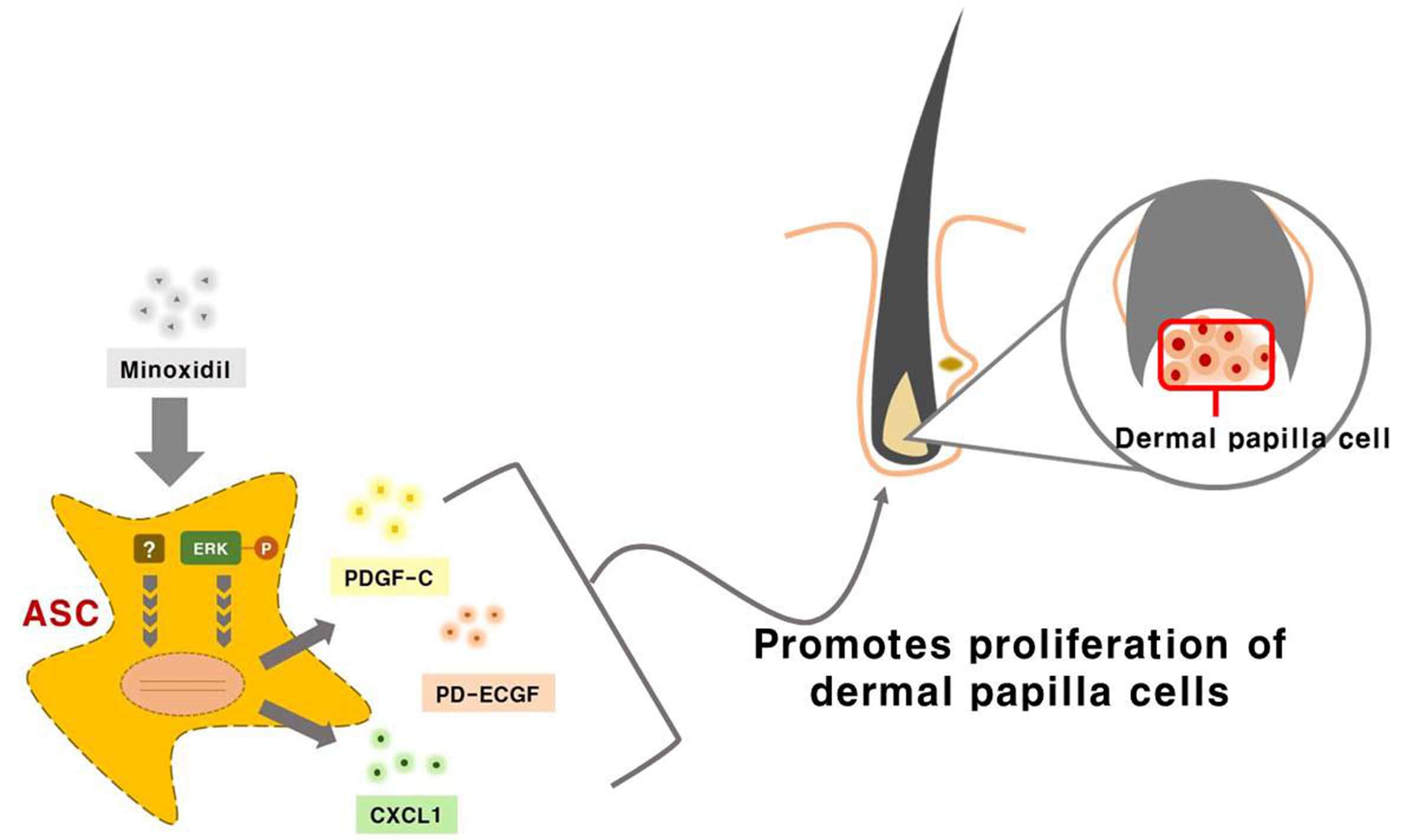{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
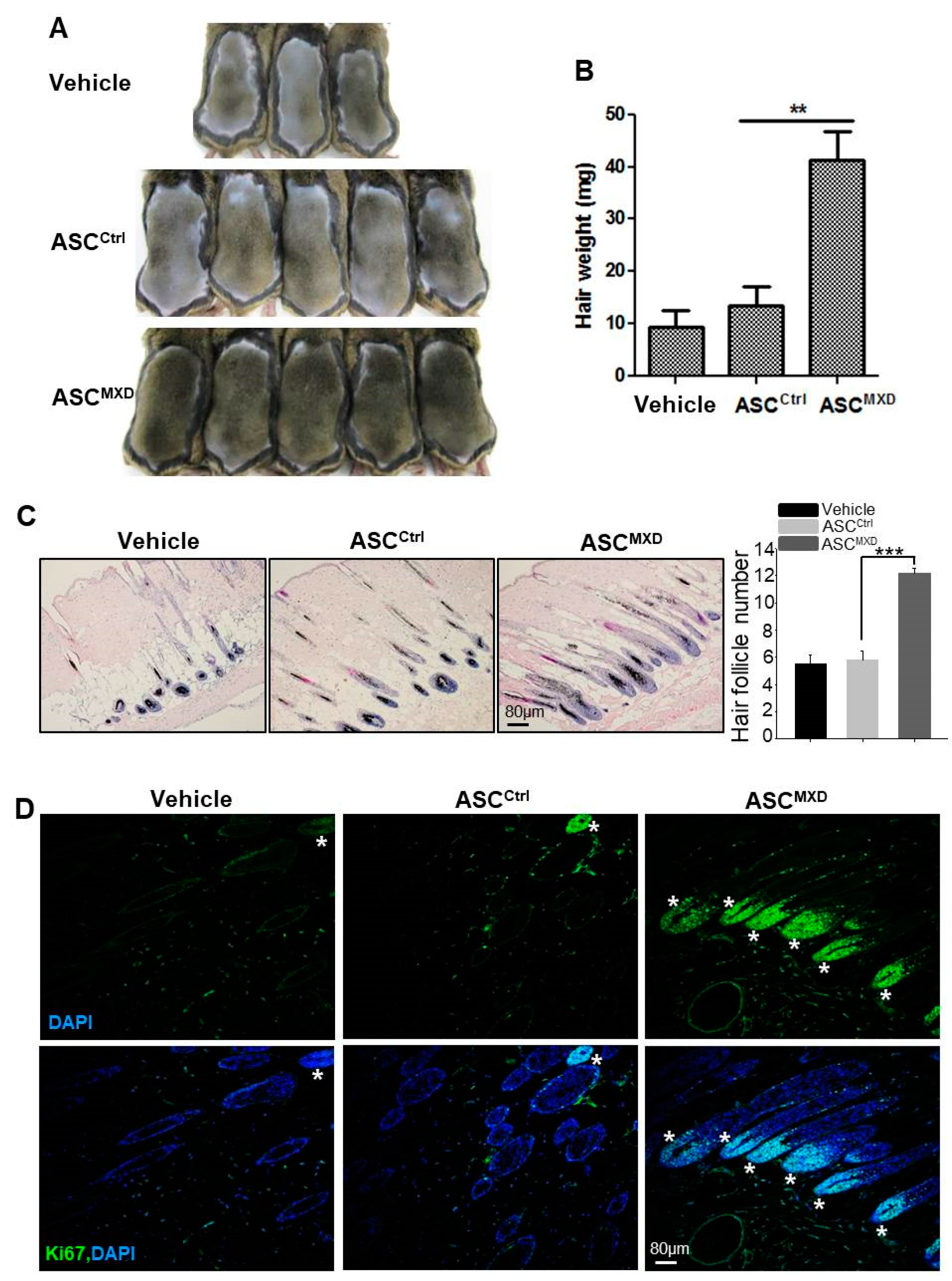
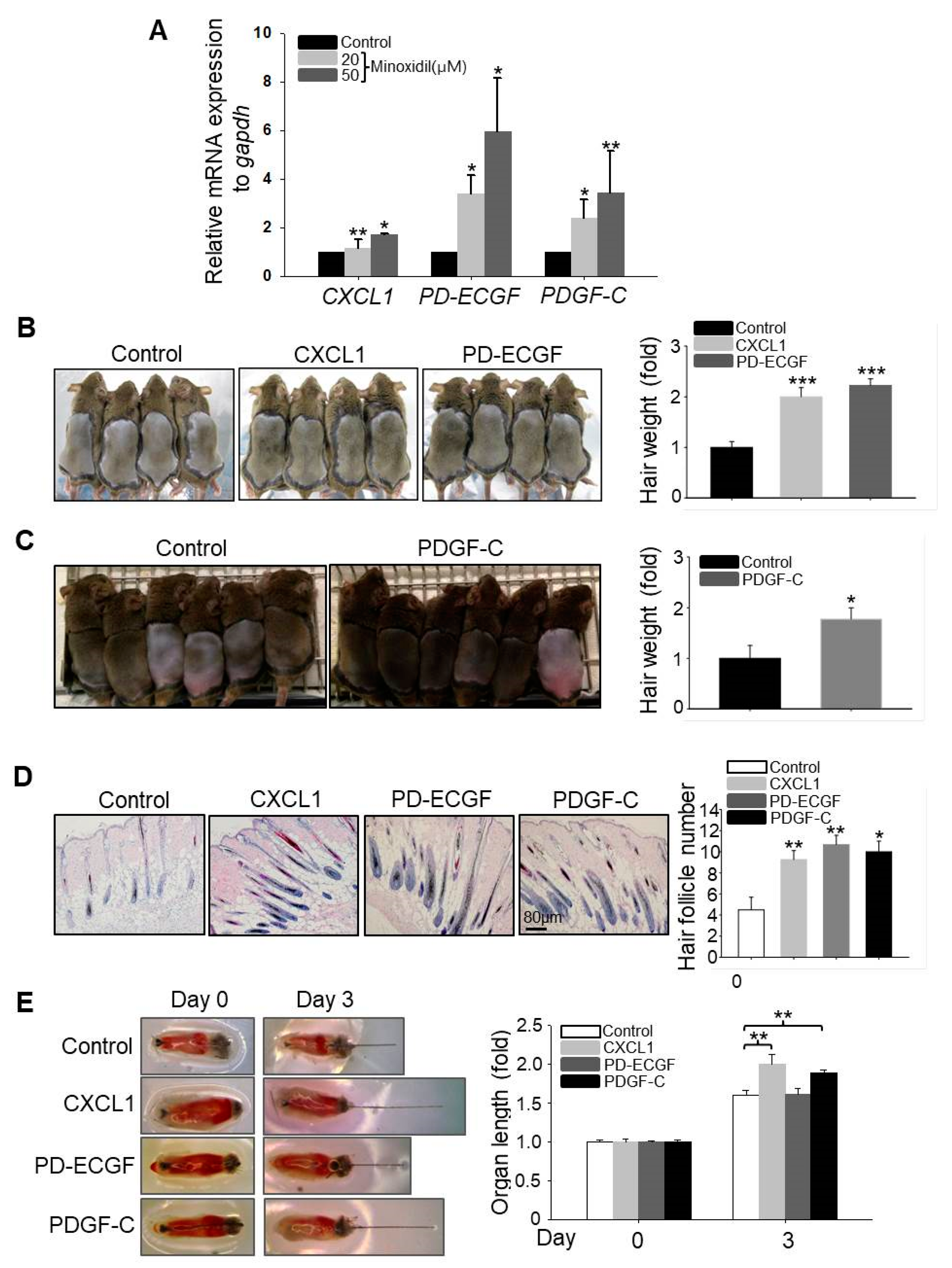
2.1. Minoxidil-Pretreated Adipose-Derived Stem Cells (ASCs) Promote Hair Growth In Vivo
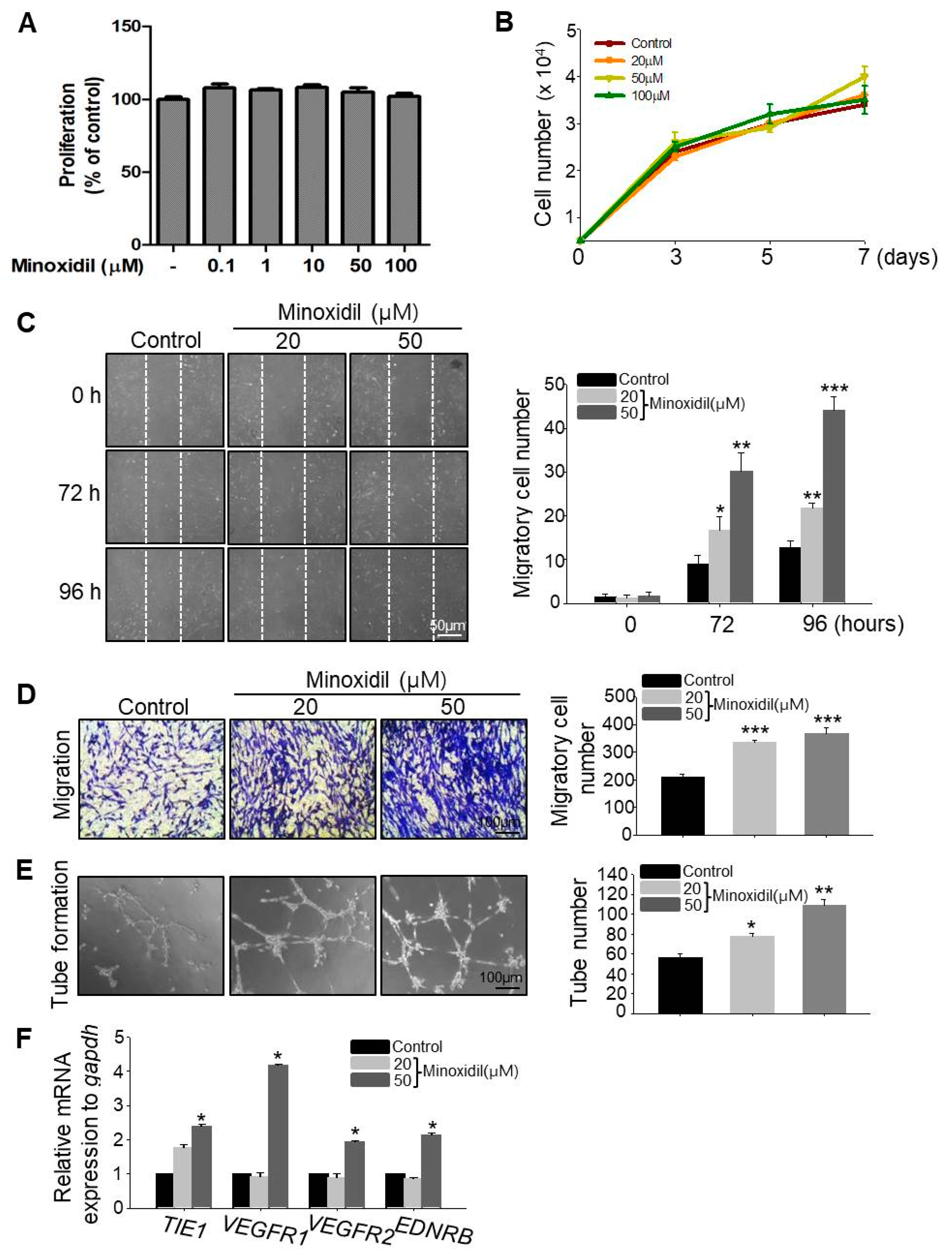
2.2. Minoxidil Can Induce Migration of ASCs
2.3. CXCL1, ECGF, and PDGF-C Induce Hair Growth
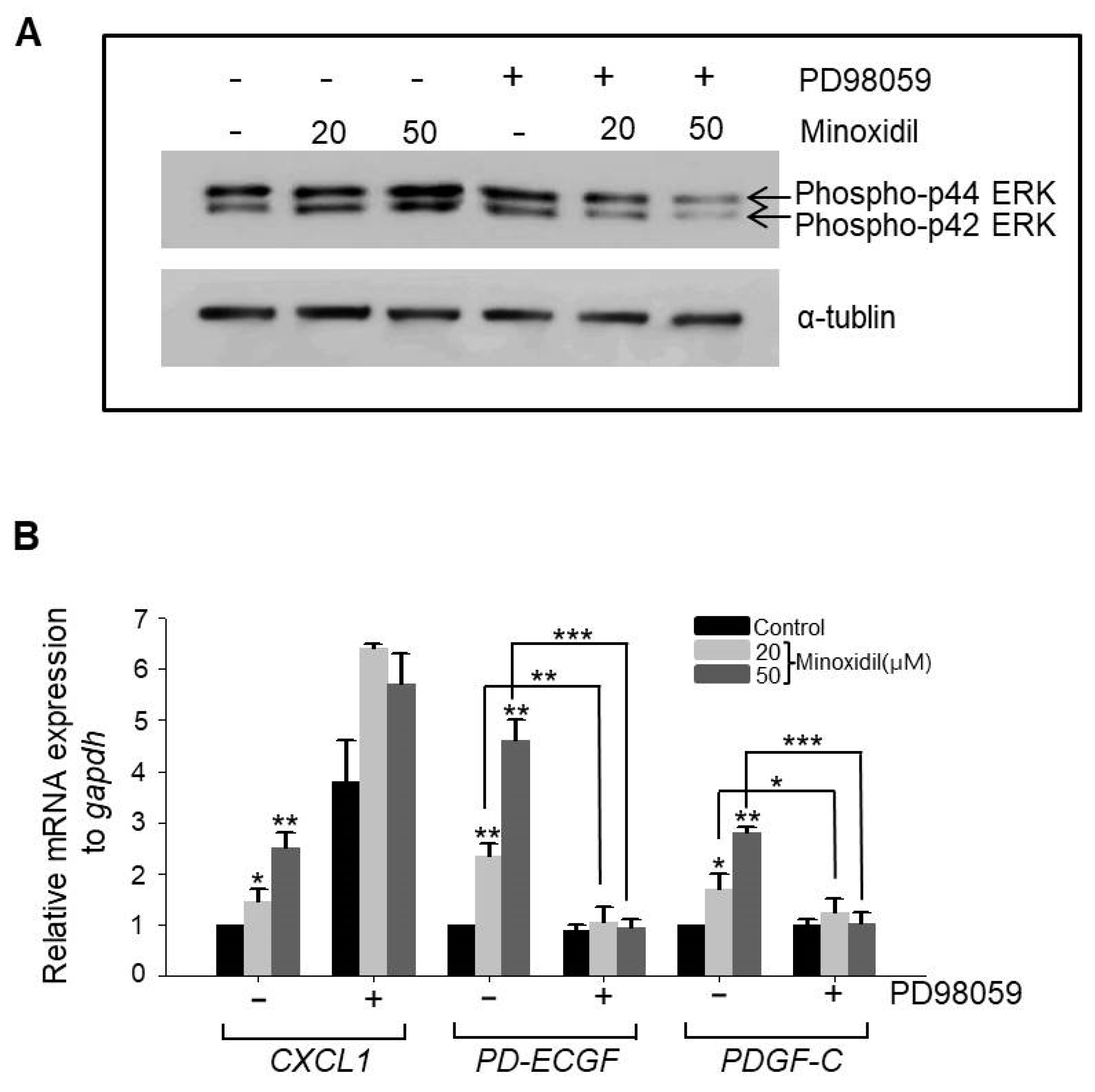
2.4. Minoxidil Regulates Expression of PD-ECGF and PDGF-C in ASCs through the ERK Pathway
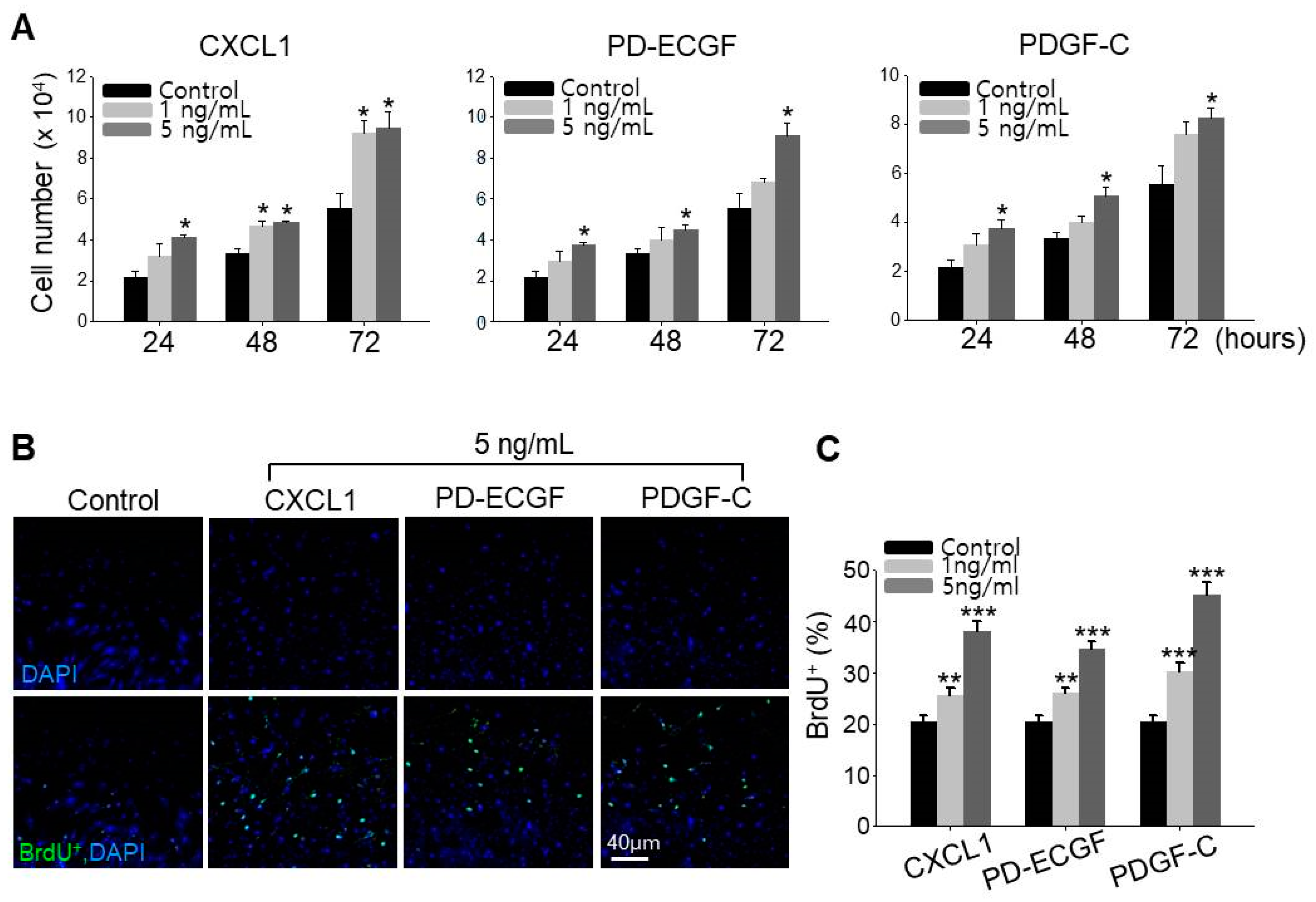
2.5. ASC Growth Factors CXCL1, PD-ECGF, and PDGF-C Induce DP Cell Proliferation
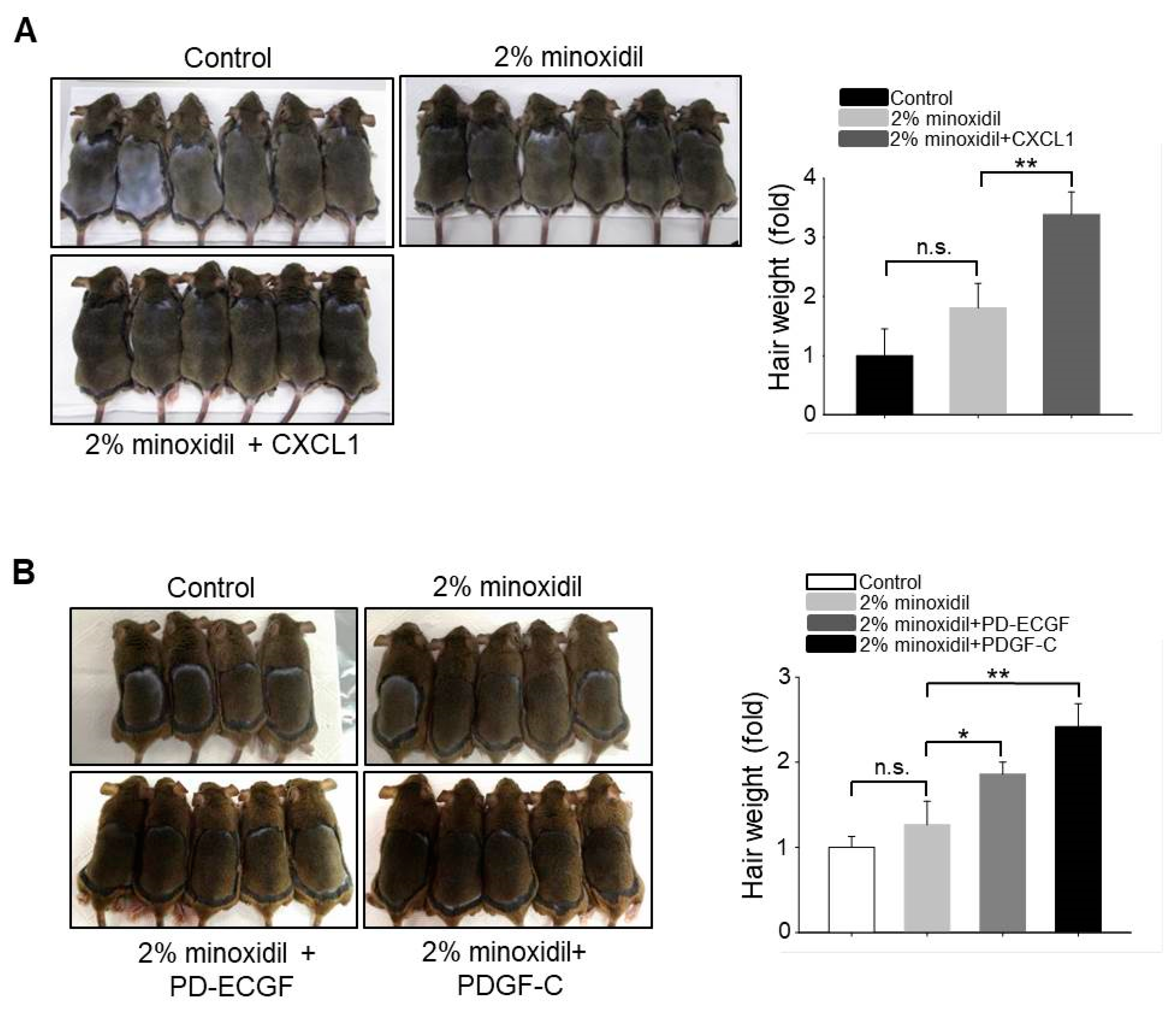
2.6. Application of CXCL1, PD-ECGF, or PDGF-C Acts Synergistically with Minoxidil to Induce Hair Growth
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Growth Assay
4.3. Scratch Migration Assay
4.4. Cell Migration Assay Using Transwell
4.5. Tube Formation Assay Using Matrigel
4.6. RNA Extraction and Quantitative RT-PCR
4.7. Western Blot
4.8. Animal Experiment
4.9. Vibrissae Follicle Organ Culture
4.10. BrdU Labeling
4.11. Hematoxylin/Eosin and Immunofluorescence Staining
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ASC | Adipose-derived stem cell |
| CXCL1 | chemokine (C-X-C motif) ligand 1 |
| PD-ECGF | platelet-derived endothelial cell growth factor |
| PDGF-C | platelet-derived growth factor-C |
References
- Won, C.H.; Yoo, H.G.; Kwon, O.S.; Sung, M.Y.; Kang, Y.J.; Chung, J.H.; Park, B.S.; Sung, J.H.; Kim, W.S.; Kim, K.H. Hair growth promoting effects of adipose tissue-derived stem cells. J. Dermatol. Sci. 2010, 57, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Festa, E.; Fretz, J.; Berry, R.; Schmidt, B.; Rodeheffer, M.; Horowitz, M.; Horsley, V. Adipocyte lineage cells contribute to the skin stem cell niche to drive hair cycling. Cell 2011, 146, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.M.; Sung, Y.K.; Kim, W.K.; Kim, J.H.; Kwack, M.H.; Yoon, I.; Kim, D.D.; Sung, J.H. Ultraviolet B preconditioning enhances the hair growth-promoting effects of adipose-derived stem cells via generation of reactive oxygen species. Stem Cells Dev. 2013, 22, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, W.K.; Sung, Y.K.; Kwack, M.H.; Song, S.Y.; Choi, J.S.; Park, S.G.; Yi, T.; Lee, H.J.; Kim, D.D.; et al. The molecular mechanism underlying the proliferating and preconditioning effect of vitamin C on adipose-derived stem cells. Stem Cells Dev. 2014, 23, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.G.; Kim, W.K.; Song, S.U.; Sung, J.H. Functional regulation of adipose-derived stem cells by PDGF-D. Stem Cells 2015, 33, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.E.; Sung, J.H. Hair regeneration using adipose-derived stem cells. Histol. Histopathol. 2016, 31, 249–256. [Google Scholar] [PubMed]
- Perez-Meza, D.; Ziering, C.; Sforza, M.; Krishnan, G.; Ball, E.; Daniels, E. Hair follicle growth by stromal vascular fraction-enhanced adipose transplantation in baldness. Stem Cells Cloning Adv. Appl. 2017, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Choi, H.; Seon, M.; Cho, D.; Bang, S.I. LL-37 stimulates the functions of adipose-derived stromal/stem cells via early growth response 1 and the MAPK pathway. Stem Cell Res. Ther. 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Buhl, A.E.; Waldon, D.J.; Kawabe, T.T.; Holland, J.M. Minoxidil stimulates mouse vibrissae follicles in organ culture. J. Dermatol. Sci. 1989, 92, 315–320. [Google Scholar] [CrossRef]
- Headington, J.T. Hair follicle biology and topical minoxidil: Possible mechanisms of action. Dermatologica 1987, 175, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Michelet, J.F.; Commo, S.; Billoni, N.; Mahe, Y.F.; Bernard, B.A. Activation of cytoprotective prostaglandin synthase-1 by minoxidil as a possible explanation for its hair growth-stimulating effect. J. Investig. Dermatol. 1997, 108, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Marubayashi, A.; Nakaya, Y.; Fukui, K.; Arase, S. Minoxidil-induced hair growth is mediated by adenosine in cultured dermal papilla cells: Possible involvement of sulfonylurea receptor 2B as a target of minoxidil. J. Investig. Dermatol. 2001, 117, 1594–1600. [Google Scholar] [PubMed]
- Kwack, M.H.; Kang, B.M.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Minoxidil activates β-catenin pathway in human dermal papilla cells: A possible explanation for its anagen prolongation effect. J. Dermatol. Sci. 2011, 62, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Otomo, S. Hair growth effect of minoxidil. Nihon Yakurigaku Zasshi. Folia Pharmacol. Jpn. 2002, 119, 167–174. [Google Scholar] [CrossRef]
- Han, J.H.; Kwon, O.S.; Chung, J.H.; Cho, K.H.; Eun, H.C.; Kim, K.H. Effect of minoxidil on proliferation and apoptosis in dermal papilla cells of human hair follicle. J. Dermatol. Sci. 2004, 34, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Liu, J.S.; Lin, A.C.; Yang, C.H.; Chung, W.H.; Wu, W.G. Minoxidil may suppress androgen receptor-related functions. Oncotarget 2014, 5, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.K.; Ahn, S.C.; Kim, D.H.; Yu, H.S. Parasite excretory-secretory proteins elicit TRIF dependent CXCL1 and IL-6 mediated allergic inflammation. Parasite Immunol. 2010, 32, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.K.; Keepers, T.R.; Gross, L.K.; Seaner, R.M.; Obrig, T.G. CXCL1/KC and CXCL2/MIP-2 are critical effectors and potential targets for therapy of Escherichia coli O157:H7-associated renal inflammation. Am. J. Pathol. 2007, 170, 526–537. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, K.; Hartmann, M.; Gunzer, A.; Roers, N.H. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, J.; Sakaguchi, H.; Hirose, R.; Tamaya, T. Expression of platelet-derived endothelial cell growth factor (PD-ECGF) related to angiogenesis in ovarian endometriosis. J. Clin. Endocrinol. Metab. 1999, 84, 359–362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Greisler, H.P.; Klosak, J.J.; Dennis, J.W.; Karesh, S.M.; Ellinger, J.; Kim, D.U. Biomaterial pretreatment with ECGF to augment endothelial cell proliferation. J. Vasc. Surg. 1987, 5, 393–399. [Google Scholar] [CrossRef]
- Santhosh Kumar, T.R.; Krishnan, L.K. Endothelial cell growth factor (ECGF) enmeshed with fibrin matrix enhances proliferation of EC in vitro. Biomaterials 2001, 22, 2769–2776. [Google Scholar] [CrossRef]
- Rezza, A.; Sennett, R.; Tanguy, M.; Clavel, C.; Rendl, M. PDGF signalling in the dermis and in dermal condensates is dispensable for hair follicle induction and formation. Exp. Dermatol. 2015, 24, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Akiyama, M.; Shimizu, H. PDGF isoforms induce and maintain anagen phase of murine hair follicles. J. Dermatol. Sci. 2006, 43, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Crisostomo, P.R.; Wang, M.; Markel, T.A.; Novotny, N.M.; Meldrum, D.R. TGF-α increases human mesenchymal stem cell-secreted VEGF by MEK- and PI3-K- but not JNK- or ERK-dependent mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1115–R1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Abarbanell, A.M.; Weil, B.R.; Herrmann, J.L.; Tan, J.; Novotny, N.M.; Coffey, A.C.; Meldrum, D.R. MEK mediates the novel cross talk between TNFR2 and TGF-EGFR in enhancing vascular endothelial growth factor (VEGF) secretion from human mesenchymal stem cells. Surgery 2009, 146, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Weil, B.R.; Herrmann, J.L.; Abarbanell, A.M.; Tan, J.; Markel, T.A.; Kelly, M.L.; Meldrum, D.R. MEK, p38, and PI-3K mediate cross talk between EGFR and TNFR in enhancing hepatocyte growth factor production from human mesenchymal stem cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1284–C1293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crisostomo, P.R.; Wang, Y.; Markel, T.A.; Wang, M.; Lahm, T.; Meldrum, D.R. Human mesenchymal stem cells stimulated by TNF-α, LPS, or hypoxia produce growth factors by an NFκB- but not JNK-dependent mechanism. Am. J. Physiol. Cell Physiol. 2008, 294, C675–682. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Pyo, H.K.; Oh, Y.J.; Han, J.H.; Lee, S.R.; Chung, J.H.; Eun, H.C.; Kim, K.H. Promotive effect of minoxidil combined with all-trans retinoic acid (tretinoin) on human hair growth in vitro. J. Korean Med. Sci. 2007, 22, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Park, B.S.; Sung, J.H.; Yang, J.M.; Park, S.B.; Kwak, S.J.; Park, J.S. Wound healing effect of adipose-derived stem cells: A critical role of secretory factors on human dermal fibroblasts. J. Dermatol. Sci. 2007, 48, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Kim, W.K.; Choi, J.S.; Song, S.Y.; Han, J.; Kim, J.H.; Kim, W.S.; Park, S.G.; Lee, H.J.; Cho, Y.K.; et al. Isolation of adipose-derived stem cells by using a subfractionation culturing method. Expert Opin. Biol. Ther. 2014, 14, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.G.; Song, S.Y.; Kim, J.K.; Sung, J.H. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adiposederived stem cells via PTPN. Cell Death Dis. 2013, 4, e588. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, S.H.; Song, S.Y.; Kim, W.S.; Song, S.U.; Yi, T.; Jeon, M.S.; Chung, H.M.; Xia, Y.; Sung, J.H. Hypoxia induces adipocyte differentiation of adiposederived stem cells by triggering reactive oxygen species generation. Cell Biol. Int. 2014, 38, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Song, S.U.; Kim, C.S.; Yoon, S.P.; Kim, S.K.; Lee, M.H.; Kang, J.S.; Choi, G.S.; Moon, S.H.; Choi, M.S.; Cho, Y.K.; et al. Variations of clonal marrow stem cell lines established from human bone marrow in surface epitopes, differentiation potential, gene expression, and cytokine secretion. Stem Cells Dev. 2008, 17, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; An, H.S.; Jeong, J.H.; Shin, S.; Song, S.Y. Megestrol Acetate Increases the Proliferation, Migration, and Adipogenic Differentiation of Adipose-Derived Stem Cells via Glucocorticoid Receptor. Stem Cells Transl. Med. 2015, 4, 789–799. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, N.; Shin, S.; Song, S.U.; Sung, J.-H. Minoxidil Promotes Hair Growth through Stimulation of Growth Factor Release from Adipose-Derived Stem Cells. Int. J. Mol. Sci. 2018, 19, 691. https://doi.org/10.3390/ijms19030691
Choi N, Shin S, Song SU, Sung J-H. Minoxidil Promotes Hair Growth through Stimulation of Growth Factor Release from Adipose-Derived Stem Cells. International Journal of Molecular Sciences. 2018; 19(3):691. https://doi.org/10.3390/ijms19030691
Chicago/Turabian StyleChoi, Nahyun, Soyoung Shin, Sun U. Song, and Jong-Hyuk Sung. 2018. "Minoxidil Promotes Hair Growth through Stimulation of Growth Factor Release from Adipose-Derived Stem Cells" International Journal of Molecular Sciences 19, no. 3: 691. https://doi.org/10.3390/ijms19030691
APA StyleChoi, N., Shin, S., Song, S. U., & Sung, J.-H. (2018). Minoxidil Promotes Hair Growth through Stimulation of Growth Factor Release from Adipose-Derived Stem Cells. International Journal of Molecular Sciences, 19(3), 691. https://doi.org/10.3390/ijms19030691