Ion Channel Disorders and Sudden Cardiac Death


Abstract
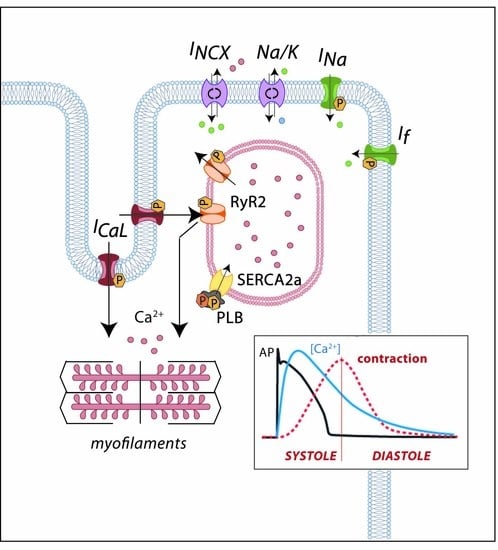
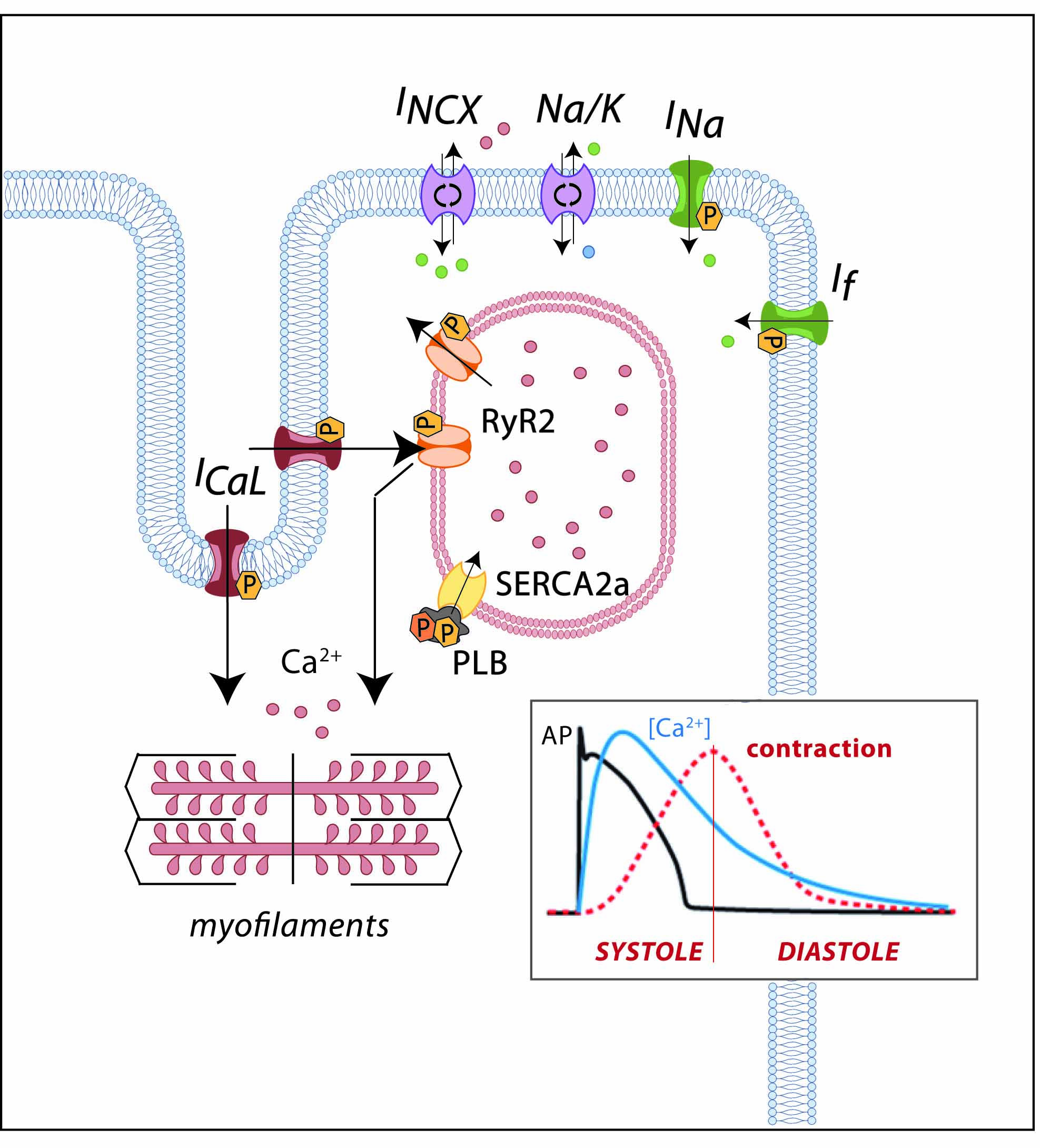
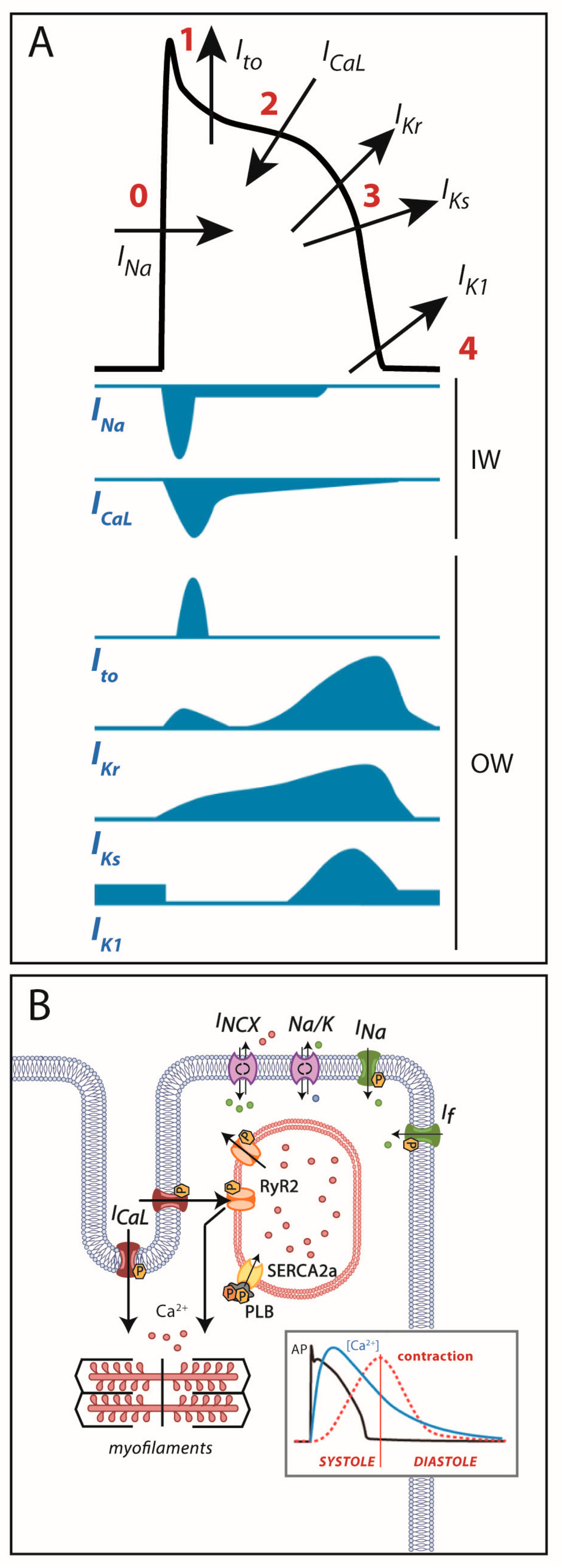
1. Introduction
2. Cardiac Electrical Physiology: Role of Ion Channels and Receptors
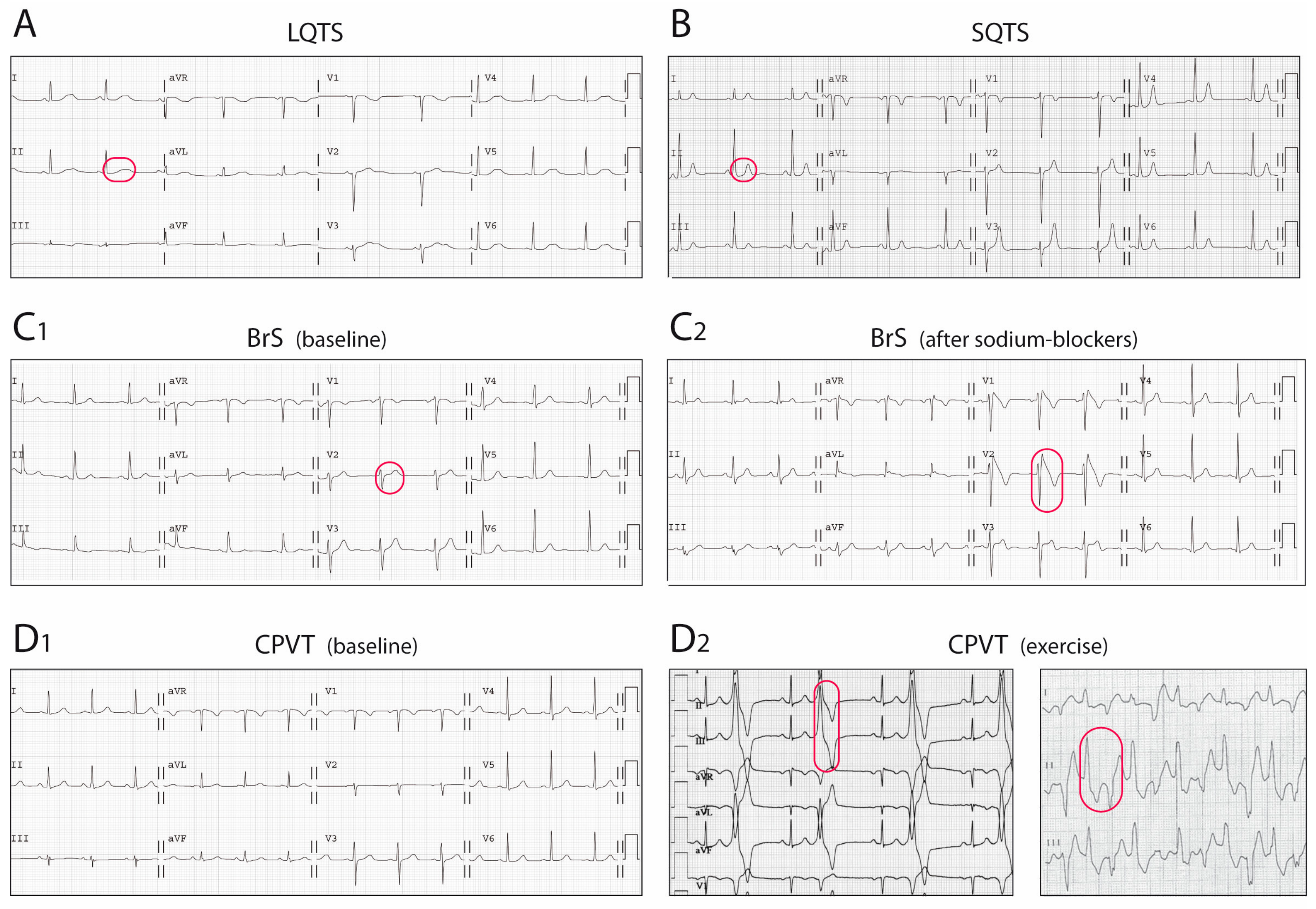
3. Long QT Syndrome
3.1. Clinical Features
3.2. Genetic Bases
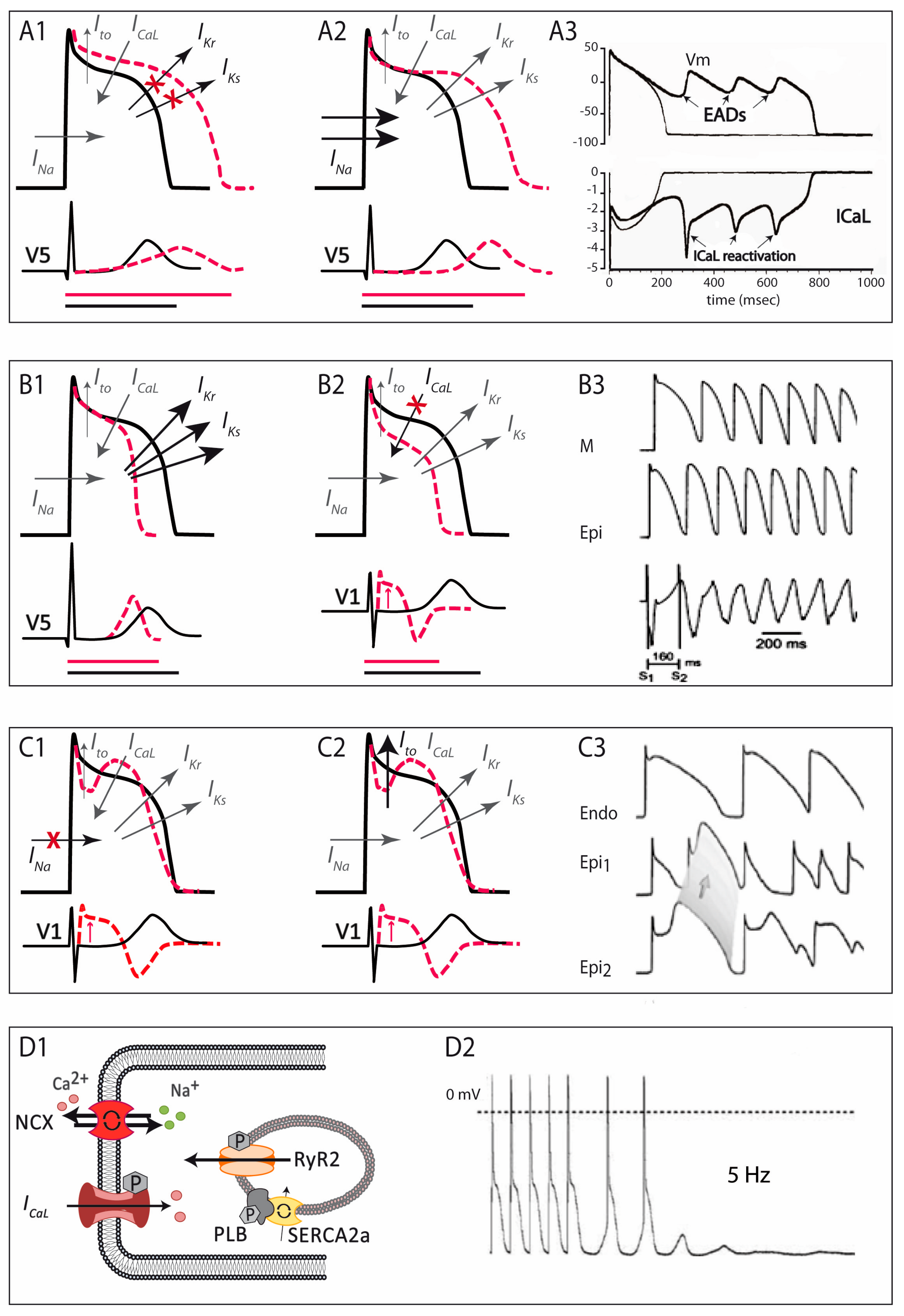
- -
- Uncommon LQTS mutations causing a decrease in outward currents. Two genes encoding regulatory β-subunits of K+ channels have been associated with the LQTS: mutations in KCNE1, encoding minK, the β1-subunit of voltage-dependent K+ channels, have been reported to interfere with the traffic of KV7.1 and lead to a reduction of IKs current [31,32]; on the other hand, mutations in KCNE2, encoding MiRP1 (minK-related peptide 1), have been reported to impair KV11.1 kinetics (slower activation, faster deactivation and increased drug sensitivity), inducing a decreased IKr [33]. Among the inward-rectifying K+ channel family, two members have also been associated to LQTS, KCNJ2 and KCNJ5. KCNJ2 encodes Kir2.1, the inward rectifying K+ channel that mediates the IK1 current [34]. Loss-of-function mutations in this gene lead not only to LQTS and susceptibility to arrhythmias, but also to periodic paralysis and developmental abnormalities, a condition known as the Andersen–Tawil syndrome [35]. KCNJ5 encodes Kir3.4, the inward rectifying K+ channel mediator of the acetylcholine/adenosine-induced IK,Ach current. Loss-of-function mutations in this gene lead to LQTS by altering the traffic of the channel [36,37]. Finally, mutations in AKAP, a gene that encodes an auxiliary protein (AKAP9, A-kinase anchor protein-9) and not an ion channel, may also cause LQTS by reducing outward currents. AKAP9 is a scaffolding protein between KV7.1 and PKA. Mutations in AKAP have been shown to reduce the KV7.1/PKA interaction, resulting in a net decrease of IKs [38].
- -
- Uncommon LQTS mutations causing an increase in inward currents. Mutations in two different β-subunits regulators of the NaV1.5 channel have been found in families with LQTS: the β1 (encoded by the SCN1B gene) and the β4 (encoded by the SCN4B gene), inducing in both cases an increased INa [39,40]. Increased ICaL currents are seen in LQTS-associated mutations on the CACNA1C gene encoding CaV1.2, the α1C-subunit of the LTCC [41]. Specifically, mutations in this gene have also been associated with the Timothy syndrome, characterized by long QT and associated serious developmental and physical disorders such as autism or immune deficiencies [42]. This particular subtype (also known as LQT8) is considered the most severe variant of LQTS, with the highest mortality rate, generally caused by extracardiac complications [19]. Other genes encoding auxiliary proteins have been associated with LQTS. ANK2 encodes ankyrin-2, a protein in charge of the assembly of the Na+/K+ exchanger, the Na+/Ca2+ exchanger and the inositol triphosphate receptor (IP3R), among others. Loss-of-function mutations in ANK2 increase ICaL by decreasing the amount of Na+/Ca2+ exchanger in the membrane leading to an abnormal restoration of the initial ion state [43,44]. All three calmodulin-encoding genes (CALM1, CALM2, and CALM3) have also been linked to LQTS. Calmodulin is an essential intracellular Ca2+ sensor that acts as a signal-transducing protein and modulates CaV1.2 (and others). Mutations in one of these genes, even in heterozygosis (meaning that only 1/6 alleles is affected), is sufficient to lead to an early and severe form of LQTS with extreme long QTc interval secondary to an impaired CaV1.2 inactivation and increased ICaL currents [45,46,47,48,49]. Mutations in the SNTA1 gene has also been associated to LQTS [50]. This gene encodes α1-syntrophin, a scaffolding protein that associates NaV1.5 channels with the nitric oxide synthase-ATPase plasma membrane Ca2+-transporting four-protein complex (NOS-PMCA4b). SNTA1 mutations disrupt this interaction and increase late INa currents [51,52]. Finally, mutations in the TRDN gene that encodes the triadin, a regulator of RyR, have been associated with the LQTS by putatively decreasing CaV1.2 inactivation and increasing ICaL currents [18].
4. Short QT Syndrome
4.1. Clinical Features
4.2. Genetic Bases
5. Brugada Syndrome
5.1. Clinical Features
5.2. Genetic Bases
6. Catecholaminergic Polymorphic Ventricular Tachycardia
6.1. Clinical Features
6.2. Genetic Bases
7. Conclusions
Acknowledgments
Authors Contribution
Conflicts of Interest
References
- Zipes, D.P.; Wellens, H.J.J. Clinical Cardiology: New Frontiers Sudden Cardiac Death. Circulation 1998, 98, 2334–2351. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.I.; Chugh, S.; DiMarco, J.; Albert, C.; Anderson, M.; Bonow, R.; Buxton, A.; Chen, P.-S.; Estes, M.; Jouven, X.; et al. Sudden Cardiac Death Prediction and Prevention Report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation 2010, 122, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Van Der Werf, C.; van Langen, I.M.; Wilde, A.A.M. Sudden death in the young: What do we know about it and how to prevent? Circ. Arrhythm. Electrophysiol. 2010, 3, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Eckart, R.E.; Shry, E.A.; Burke, A.P.; McNear, J.A.; Appel, D.A.; Castillo-Rojas, L.M.; Avedissian, L.; Pearse, L.A.; Potter, R.N.; Tremaine, L.; et al. Sudden death in young adults: An autopsy-based series of a population undergoing active surveillance. J. Am. Coll. Cardiol. 2011, 58, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Puranik, R.; Chow, C.K.; Duflou, J.A.; Kilborn, M.J.; McGuire, M.A. Sudden death in the young. Heart Rhythm 2005, 2, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Benito, B. Sudden death in patients without structural heart disease. Rev. Española Cardiol. 2013, 13, 14–23. [Google Scholar]
- Cheng, Y.; Lin, X.; Ji, C.; Chen, X.; Liu, L.; Tang, K.; Wu, S. Role of Early Repolarization Pattern in Increasing Risk of Death. J. Am. Heart Assoc. 2016, 5, e003375. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Underwood, K.; Nevelev, D.; Kofman, S.; Priori, S.G. The new kids on the block of arrhythmogenic disorders: Short QT syndrome and early repolarization. J. Cardiovasc. Electrophysiol. 2017, 28, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Ackerman, M.J. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 2011, 123, 1021–1037. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Gillis, A.M.; Bryant, W.J.; Hlatky, M.A.; Callans, D.J.; Granger, C.B.; Curtis, A.B.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary. Heart Rhythm 2017, 24390. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Ackerman, M.J. Exercise extreme caution when calling rare genetic variants novel arrhythmia syndrome susceptibility mutations. Heart Rhythm 2010, 7, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; Mosca, F.; et al. Prevalence of the congenital long-qt syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Current Perspectives Diagnostic Criteria for the Long QT Syndrome An Update. Circulation 1993, 782–785. [Google Scholar] [CrossRef]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk stratification in the long-QT syndrome. N. Engl. J. Med. 2003, 348, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J. Genotype-phenotype correlation in the long-QT syndrome. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Moss, A.J.; Peterson, D.R.; McNitt, S.; Zareba, W.; Andrews, M.L.; Robinson, J.L.; Locati, E.H.; Ackerman, M.J.; Benhorin, J.; et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital Long-QT syndrome. Circulation 2008, 117, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Altmann, H.M.; Tester, D.J.; Will, M.L.; Middha, S.; Evans, J.M.; Eckloff, B.W.; Ackerman, M.J. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: Elucidation of the triadin knockout syndrome. Circulation 2015, 131, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Lahrouchi, N.; Priori, S.G. Genetics of Sudden Cardiac Death. Circ. Res. 2015, 116, 1919–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Oliveras, A.; Bartolucci, C.; Muñoz, C.; de la Cruz, A.; Peraza, D.A.; Gimeno, J.R.; Martín-Martínez, M.; Severi, S.; Felipe, A.; et al. D242N, a KV7.1 LQTS mutation uncovers a key residue for IKsvoltage dependence. J. Mol. Cell. Cardiol. 2017, 110, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Thottathil, P.; Lopes, C.M.; Moss, A.J.; McNitt, S.; Jin, O.U.; Robinson, J.L.; Zareba, W.; Ackerman, M.J.; Kaufman, E.S.; et al. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm 2012, 9, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Shimizu, W.; Wilde, A.A.M.; Towbin, J.A.; Zareba, W.; Robinson, J.L.; Qi, M.; Vincent, G.M.; Ackerman, M.J.; Kaufman, E.S.; et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007, 115, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Furutani, M.; Trudeau, M.; Hagiwara, N.; Seki, A.; Gong, Q.; Zhou, Z.; Imamura, S.; Nagashima, H.; Kasanuki, H.; Takao, A.; et al. Novel Mechanism Associated With an Inherited Cardiac Arrhythmia. Circulation 1999, 99, 2290–2295. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Anderson, C.L.; Burgess, D.E.; Elayi, C.S.; January, C.T.; Delisle, B.P. Molecular pathogenesis of long QT syndrome type 2. J. Arrhythmia 2016, 32, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Zareba, W.; Kaufman, E.S.; Gartman, E.; Peterson, J.; Benhorin, D.R.; Towbin, J.A.; Keating, M.T.; Priori, S.G.; Schwartz, P.J.; et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002, 105, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, W.; Moss, A.J.; Wilde, A.A.M.; Towbin, J.A.; Ackerman, M.J.; January, C.T.; Tester, D.J.; Zareba, W.; Robinson, J.L.; Qi, M.; et al. Genotype-Phenotype Aspects of Type 2 Long QT Syndrome. J. Am. Coll. Cardiol. 2009, 54, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Wilde, A.; Moss, A.; Kaufman, E.; Shimizu, W.; Peterson, D.; Benhorin, J.; Lopes, C.; Towbin, J.; Spazzolini, C.; Crotti, L.; et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 2016, 134, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Krumerman, A.; Gao, X.; Bian, J.; Melman, Y.F.; Kagan, A.; Mcdonald, T.V.; Gao, X.; Bian, J.; Melman, F.; Kagan, A.; et al. An LQT mutant minK alters KvLQT1 trafficking Andrew. Am. J. Physiol. Cell Physiol. 2004, 10461, 1453–1464. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harmer, S.C.; Tinker, A. The role of abnormal trafficking of KCNE1 in long QT syndrome 5. Biochem. Soc. Trans. 2007, 35, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef]
- Fodstad, H.; Swan, H.; Auberson, M.; Gautschi, I.; Loffing, J.; Schild, L.; Kontula, K. Loss-of-function mutations of the K+channel gene KCNJ2 constitute a rare cause of long QT syndrome. J. Mol. Cell. Cardiol. 2004, 37, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Ptacek, L.J.; Pavlakis, S.G.; DeVivo, D.C.; Penn, A.S.; Özdemir, C.; Griggs, R.C. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994, 35, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; et al. Identification of a Kir3.4 Mutation in Congenital Long QT Syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, J.; Hong, L.; Liang, B.; Graff, C.; Yang, Y.; Christiansen, M.; Olesen, S.P.; Zhang, L.; Kanters, J.K. The phenotype characteristics of type 13 long QT syndrome with mutation in KCNJ5 (Kir3.4-G387R). Heart Rhythm 2013, 10, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef] [PubMed]
- Riuró, H.; Campuzano, O.; Arbelo, E.; Iglesias, A.; Batlle, M.; Pérez-Villa, F.; Brugada, J.; Pérez, G.J.; Scornik, F.S.; Brugada, R. A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 2014, 11, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Domingo, A.; Kaku, T.; Tester, D.J.; Iturralde-Torres, P.; Itty, A.; Ye, B.; Valdivia, C.; Ueda, K.; Canizales-Quinteros, S.; Tusié-Luna, M.T.; et al. SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation 2007, 116, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Boczek, N.J.; Ye, D.; Miyake, C.Y.; De la Uz, C.M.; Allen, H.D.; Ackerman, M.J.; Kim, J.J. Novel long QT syndrome-associated missense mutation, L762F, in CACNA1C-encoded L-type calcium channel imparts a slower inactivation tau and increased sustained and window current. Int. J. Cardiol. 2016, 220, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Schott, J.-J.; Gramolini, A.O.; Dilly, K.W.; Guatimoisim, S.; duBell, W.H.; Song, L.-S.; Haurogne, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-B mutations causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003, 421, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Swayne, L.A.; Murphy, N.P.; Asuri, S.; Chen, L.; Xu, X.; McIntosh, S.; Wang, C.; Lancione, P.J.; Roberts, J.D.; Kerr, C.; et al. Novel Variant in the ANK2 Membrane-Binding Domain Is Associated with Ankyrin-B Syndrome and Structural Heart Disease in a First Nations Population with a High Rate of Long QT Syndrome. Circ. Cardiovasc. Genet. 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Johnson, C.N.; Graf, E.; de Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.J.; Boczek, N.J.; Etheridge, S.P.; Ackerman, M.J. CALM3 mutation associated with long QT syndrome. Heart Rhythm 2015, 12, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Gomez-Hurtado, N.; Ye, D.; Calvert, M.L.; Tester, D.J.; Kryshtal, D.O.; Hwang, H.S.; Johnson, C.N.; Chazin, W.J.; Loporcaro, C.G.; et al. Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ. Cardiovasc. Genet. 2016, 9, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Wang, C.; Thomas, M.J.; Pitt, G.S. α1-Syntrophin variant identified in drug-induced long QT syndrome increases late sodium current. PLoS ONE 2016, 11, e0152355. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ai, T.; Kim, J.J.; Mohapatra, B.; Xi, Y.; Li, Z.; Abbasi, S.; Purevjav, E.; Samani, K.; Ackerman, M.J.; et al. Alpha-1-syntrophin mutation and the long-QT syndrome: A disease of sodium channel disruption. Circ. Arrhythm. Electrophysiol. 2008, 1, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, R.; Vega, A.L.; Song, C.; Zhou, Q.; Tan, B.; Berger, S.; Makielski, J.C.; Eckhardt, L.L. The interaction of caveolin 3 protein with the potassium inward rectifier channel Kir2.1: Physiology and pathology related to long QT syndrome 9 (LQT9). J. Biol. Chem. 2013, 288, 17472–17480. [Google Scholar] [CrossRef] [PubMed]
- Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepalli, R.C.; Foell, J.D.; Li, Z.; et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 2006, 114, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Hedley, P.L.; Kanters, J.K.; Dembic, M.; Jespersen, T.; Skibsbye, L.; Aidt, F.H.; Eschen, O.; Graff, C.; Behr, E.R.; Schlamowitz, S.; et al. The role of CAV3 in long-QT syndrome: Clinical and functional assessment of a caveolin-3/Kv11.1 double heterozygote versus caveolin-3 single heterozygote. Circ. Cardiovasc. Genet. 2013, 6, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Hof, T.; Liu, H.; Salle, L.; Schott, J.; Ducreux, C.; Millat, G.; Chevalier, P.; Probst, V.; Guinamard, R.; Bouvagnet, P.; et al. TRPM4 non-selective cation channel variants in long QT syndrome. BMC Med. Genet. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Kiehne, N.; Erkapic, D.; Schmidt, J.; Hamm, C.W.; Bratzke, H.; Pitschner, H.F.; Kuniss, M.; Neumann, T. A novel mutation in the cardiac ryanodine receptor gene (RyR2) in a patient with an unequivocal LQTS. Int. J. Cardiol. 2011, 146, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Ichikawa, M.; Takayama, K.; Fukumoto, D.; Horie, M. Novel RYR2 mutations causative for long QT syndromes. Eur. Heart J. 2017, 38, 1–28. [Google Scholar] [CrossRef][Green Version]
- Tester, D.J.; Kopplin, L.J.; Will, M.L.; Ackerman, M.J. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic Short QT Interval: A New Clinical Syndrome? Cardiology 2000, 94, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Gaita, F.; Giustetto, C.; Bianchi, F.; Wolpert, C.; Schimpf, R.; Riccardi, R.; Grossi, S.; Richiardi, E.; Borggrefe, M. Short QT syndrome: A familial cause of sudden death. Circulation 2003, 108, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, C.; Di Monte, F.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Sbragia, P.; Leone, G.; Maury, P.; Anttonen, O.; Haissaguerre, M.; et al. Short QT syndrome: Clinical findings and diagnostic-therapeutic implications. Eur. Heart J. 2006, 27, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Kanthan, A.; Monteforte, N.; Memmi, M.; Bloise, R.; Novelli, V.; Miceli, C.; O’Rourke, S.; Borio, G.; Zienciuk-Krajka, A.; et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 2014, 63, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Saguner, A.M.; Medeiros-Domingo, A.; Schaller, A.; Balmer, C.; Steffel, J.; Brunckhorst, C.; Duru, F. Multiple clinical profiles of families with the short QT syndrome. Europace 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Brugada, R.; Hong, K.; Dumaine, R.; Cordeiro, J.; Gaita, F.; Borggrefe, M.; Menendez, T.M.; Brugada, J.; Pollevick, G.D.; Wolpert, C.; et al. Sudden Death Associated with Short-QT Syndrome Linked to Mutations in HERG. Circulation 2004, 109, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Sakaguchi, T.; Ashihara, T.; Ding, W.G.; Nagaoka, I.; Oka, Y.; Nakazawa, Y.; Yao, T.; Jo, H.; Ito, M.; et al. A novel KCNH2 mutation as a modifier for short QT interval. Int. J. Cardiol. 2009, 137, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Quan, X.Q.; Fromme, S.; Cox, R.H.; Zhang, P.; Zhang, L.; Guo, D.; Guo, J.; Patel, C.; Kowey, P.R.; et al. A novel mutation in the KCNH2 gene associated with short QT syndrome. J. Mol. Cell. Cardiol. 2011, 50, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Bellocq, C.; van Ginneken, A.C.G.; Bezzina, C.R.; Alders, M.; Escande, D.; Mannens, M.M.A.M.; Baró, I.; Wilde, A.A.M. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004, 109, 2394–2397. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Oliveras, A.; de La Cruz, A.; Bartolucci, C.; Muñoz, C.; Salar, E.; Gimeno, J.R.; Severi, S.; Comes, N.; Felipe, A.; et al. A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc. Res. 2015, 107, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; di Barletta, M.R.; Gudapakkam, S.; et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005, 96, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Makiyama, T.; Akao, M.; Ehara, E.; Ohno, S.; Iguchi, M.; Nishio, Y.; Sasaki, K.; Itoh, H.; Yokode, M.; et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc. Res. 2012, 93, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, E.; Sicca, F.; Brignone, M.S.; D’adamo, M.C.; Napolitano, C.; Servettini, I.; Moro, F.; Ruan, Y.; Guglielmi, L.; Pieroni, S.; et al. Genetically induced dysfunctions of Kir2.1 channels: Implications for short QT3 syndrome and autism-epilepsy phenotype. Hum. Mol. Genet. 2014, 23, 4875–4886. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Ghadri, J.R.; Rougier, J.S.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D.; et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Benito, B.; Brugada, J.; Brugada, R.; Brugada, P. Brugada Syndrome. Rev. Española Cardiol. 2009, 62, 1297–1315. [Google Scholar] [CrossRef]
- Park, D.S.; Cerrone, M.; Morley, G.; Vasquez, C.; Fowler, S.; Liu, N.; Bernstein, S.A.; Liu, F.Y.; Zhang, J.; Rogers, C.S.; et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J. Clin. Investig. 2015, 125, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Brugada, R.; Brugada, J.; Brugada, P. Brugada Syndrome. Prog. Cardiovasc. Dis. 2008, 51, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Baranchuk, A.; Nguyen, T.; Ryu, M.H.; Femenía, F.; Zareba, W.; Wilde, A.A.M.; Shimizu, W.; Brugada, P.; Pérez-Riera, A.R. Brugada phenocopy: New terminology and proposed classification. Ann. Noninvasive Electrocardiol. 2012, 17, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Perez Riera, A.R.; et al. Brugada syndrome: Report of the second consensus conference. Heart Rhythm 2005, 2, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Giordano, U.; Bloise, R.; Giustetto, C.; de Nardis, R.; Grillo, M.; et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation 2002, 105, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Brugada, R.; Antzelevitch, C.; Towbin, J.; Nademanee, K.; Brugada, P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1to V3. Circulation 2002, 105, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender Differences in Clinical Manifestations of Brugada Syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Sroubek, J.; Probst, V.; Mazzanti, A.; Hevia, J.C.; Ohkubo, K.; Zorzi, A.; Kostopoulou, A.; Yin, X.; Napolitano, C.; Milan, D.J.; et al. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation 2016, 133, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Pappone, C.; Berruezo, A.; Vicedomini, G.; Manguso, F.; Ciconte, G.; Giannelli, L.; Santinelli, V. Brugada Syndrome Phenotype Elimination by Epicardial Substrate Ablation. Circ. Arrhythm. Electrophysiol. 2015, 8, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Juang, J.M.J.; Horie, M. Genetics of Brugada syndrome. J. Arrhythmia 2016, 32, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Brignole, M.; Giordano, U.; Giovannini, T.; Menozzi, C.; Bloise, R.; et al. Clinical and Genetic Heterogeneity of Right Bundle Branch Block and ST-Segment Elevation Syndrome. Circulation 2000, 102, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; Kamakura, S.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, P.G.; Tan, H.L.; Probst, V.; Koopmann, T.T.; Tanck, M.W.; Bhuiyan, Z.A.; Sacher, F.; Kyndt, F.; Schott, J.J.; Albuisson, J.; et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009, 6, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Campuzano, O.; Ishac, R.; Iglesias, A.; Junttila, M.; Michaud, J.; Brugada, J.; Brugada, P.; Brugada, R. Role of genetic testing in risk stratification of Brugada syndrome. Eur. Heart J. 2009, 30, 5121. [Google Scholar]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. Structure 2008, 118, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Riuró, H.; Beltran-Alvarez, P.; Tarradas, A.; Selga, E.; Campuzano, O.; Vergés, M.; Pagans, S.; Iglesias, A.; Brugada, J.; Brugada, P.; et al. A Missense Mutation in the Sodium Channel β2 Subunit Reveals SCN2B as a New Candidate Gene for Brugada Syndrome. Hum. Mutat. 2013, 34, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martinez, H.; Burashnikov, E.; Springer, M.; Wu, Y.; Varro, A.; Pfeiffer, R.; Koopmann, T.T.; Cordeiro, J.M.; Guerchicoff, A.; et al. A mutation in the β3 subunit of the cardiac sodium channel associated with brugada ECG phenotype. Circ. Cardiovasc. Genet. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Takahashi, N.; Ohno, S.; Sakurada, H.; Nakamura, K.; On, Y.K.; Park, J.E.; Makiyama, T.; Horie, M.; Arimura, T.; et al. Novel SCN3B mutation associated with brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ. J. 2013, 77, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Remme, C.A.; Schumacher, C.A.; Scicluna, B.P.; Wolswinkel, R.; de Jonge, B.; Bezzina, C.R.; Veldkamp, M.W. Functional NaV1.8 channels in intracardiac neurons: The link between SCN10A and cardiac electrophysiology. Circ. Res. 2012, 111, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Béziau, D.M.; Barc, J.; O’Hara, T.; Le Gloan, L.; Amarouch, M.Y.; Solnon, A.; Pavin, D.; Lecointe, S.; Bouillet, P.; Gourraud, J.B.; et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res. Cardiol. 2014, 109, 446. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Wang, Q.; Shirayama, T.; Itoh, H.; Horie, M. . Nonsense-mediated mRNA decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm 2014, 11, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient Outward Current (Ito) Gain-of-Function Mutations in the Transient Outward Current (Ito) KCND3-Encoded Kv4.3 Potassium Channel and Brugada Syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Mao, W.; Cai, B.; Li, F.; Xu, H. Two novel Brugada syndrome-associated mutations increase KV4.3 membrane expression and function. Int. J. Mol. Med. 2015, 36, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Radicke, S.; Cotella, D.; Graf, E.M.; Banse, U.; Jost, N.; Varró, A.; Tseng, G.N.; Ravens, U.; Wettwer, E. Functional modulation of the transient outward current Itoby KCNE β-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc. Res. 2006, 71, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Delpón, E.; Cordeiro, J.M.; Núñez, L.; Thomsen, P.E.B.; Guerchicoff, A.; Pollevick, G.D.; Wu, Y.; Kanters, J.K.; Larsen, C.T.; Hofman-Bang, J.; et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2008, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Portero, V.; Le Scouarnec, S.; Es-Salah-Lamoureux, Z.; Burel, S.; Gourraud, J.B.; Bonnaud, S.; Lindenbaum, P.; Simonet, F.; Violleau, J.; Baron, E.; et al. Dysfunction of the voltage-gated K+channel β2 subunit in a familial case of Brugada syndrome. J. Am. Heart Assoc. 2016, 5, e003122. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Adler, A.; Green, S.; Al-Zoughool, F.; Doroshenko, P.; Orr, N.; Uppal, S.; Healey, J.S.; Birnie, D.; Sanatani, S.; et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ. Cardiovasc. Genet. 2014, 7, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Zankov, P.; Ding, W.G.; Itoh, H.; Makiyama, T.; Doi, T.; Shizuta, S.; Hattori, T.; Miyamoto, A.; Naiki, N.; et al. KCNE5 (KCNE1L) variants are novel modulators of brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-domingo, A.; Tan, B.; Crotti, L.; Tester, D.J.; Eckhardt, L.; Cuoretti, A.; Kroboth, S.L.; Song, C.; Zhou, Q.; Kopp, D.; et al. Gain-of-Function Mutations, S422L, in the KCNJ8-Encoded Cardiac K-ATP Channel Kir6.1 as a Pathogenic Substrate for J Wave Syndromes. Heart Rhythm 2010, 7, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martínez, H.; Hu, D.; Ferrer, T.; Onetti, C.G.; Wu, Y.; Burashnikov, E.; Boyle, M.; Surman, T.; Urrutia, J.; Veltmann, C.; et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 2012, 9, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Wilders, R.; Verkerk, A.O. Role of the R1135H KCNH2 mutation in Brugada syndrome. Int. J. Cardiol. 2010, 144, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ohno, S.; Ding, W.G.; Fukuyama, M.; Miyamoto, A.; Itoh, H.; Makiyama, T.; Wu, J.; Bai, J.; Hasegawa, K.; et al. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2014, 25, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakamura, K.; Hayashi, T.; Inagaki, N.; Takahashi, M.; Arimura, T.; Morita, H.; Higashiuesato, Y.; Hirano, Y.; Yasunami, M.; et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 2004, 279, 27194–27198. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.; Aquila, M.; Hertel, B.; Berthold, A.; Neumann, T.; DiFrancesco, D.; Moroni, A.; Thiel, G.; Kauferstein, S. Mutation in S6 domain of HCN4 channel in patient with suspected Brugada syndrome modifies channel function. Pflugers Arch. Eur. J. Physiol. 2016, 468, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 2010, 3, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular Genetics and Functional Anomalies in a Series of 248 Brugada Cases with 11 Mutations in the TRPM4 Channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sall, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hennessey, J.A.; Kirkton, R.D.; Wang, C.; Graham, V.; Puranam, R.S.; Rosenberg, P.B.; Bursac, N.; Pitt, G.S. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 2011, 109, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, J.A.; Marcou, C.A.; Wang, C.; Wei, E.Q.; Wang, C.; Tester, D.J.; Torchio, M.; Dagradi, F.; Crotti, L.; Schwartz, P.J.; et al. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013, 10, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- London, B.; Michalec, M.; Mehdi, H.; Zhu, X.; Kerchner, L.; Sanyal, S.; Viswanathan, P.C.; Pfahnl, A.E.; Shang, L.L.; Madhusudanan, M.; et al. Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007, 116, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, Y.-Q.; Fan, L.-L.; Guo, S.; Li, J.-J.; Jin, J.-Y.; Xiang, R. Whole-exome sequencing identifies a novel mutation of GPD1L (R189X) associated with familial conduction disease and sudden death. J. Cell. Mol. Med. 2018, 22, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A novel disease gene for Brugada syndrome: Sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. MOG1: A new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.S.; Holst, A.G.; Schmitt, N. Letter by Olesen et al. regarding article “MOG1: A New susceptibility gene for brugada syndrome”. Circ. Cardiovasc. Genet. 2011, 4, 2010–2011. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Campuzano, O.; Berne, P.; Selga, E.; Allegue, C.; Iglesias, A.; Brugada, J.; Brugada, R. Brugada syndrome and p.E61X_RANGRF. Cardiol. J. 2014, 21, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Ye, D.; Johnson, E.K.; Wang, W.; Crotti, L.; Tester, D.J.; Dagradi, F.; Mizusawa, Y.; Torchio, M.; Alders, M.; et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ. Res. 2014, 115, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, L.; Crotti, P.J.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Podliesna, S.; Tadros, R.; Lodder, E.M.; Mengarelli, I.; De Jonge, B.; Beekman, L.; Barc, J.; Wilders, R.; Wilde, A.A.M.; et al. The brugada syndrome susceptibility gene HEY2 modulates cardiac transmural ion channel patterning and electrical heterogeneity. Circ. Res. 2017, 121, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.S.; Tynan, M.; Braidwood, L.; Fitzgerald, G.R. Bidirectional tachycardia in a child. A study using His bundle electrography. Br. Heart J. 1975, 37, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; do Ngoc, D.; Coumel, P. Catecholaminergic polymorphic ventricular tachycardia in children. Circulation 1995, 95, 1512–1519. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ruan, Y.; Priori, S.G. Catecholaminergic Polymorphic Ventricular Tachycardia. Prog. Cardiovasc. Dis. 2008, 51, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J. Arrhythm. 2016, 32, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Bloise, R.; Memmi, M.; Priori, S.G. Clinical utility gene card for: Catecholaminergic polymorphic ventricular tachycardia (CPVT). Eur. J. Hum. Genet. 2014, 22, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Bloise, R.; Monteforte, N.; Priori, S.G. Sudden cardiac death and genetic ion channelopathies: Long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation 2012, 125, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Moore, J.P.; Cerrone, M.; Priori, S.G.; Kertesz, N.J.; Ro, P.S.; Batra, A.S.; Kaufman, E.S.; Fairbrother, D.L.; Saarel, E.V.; et al. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia a randomized clinical trial. JAMA Cardiol. 2017, 2, 759–766. [Google Scholar] [PubMed]
- Roston, T.M.; Vinocur, J.M.; Maginot, K.R.; Mohammed, S.; Salerno, J.C.; Etheridge, S.P.; Cohen, M.; Hamilton, R.M.; Pflaumer, A.; Kanter, R.J.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia in Children: Analysis of Therapeutic Strategies and Outcomes from an International Multicenter Registry. Circ. Arrhythm. Electrophysiol. 2015, 8, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; Wilde, A.A.; van der Werf, C. The Role of Flecainide in the Management of Catecholaminergic Polymorphic Ventricular Tachycardia. Arrhythm. Electrophysiol. Rev. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Chopra, N.; Laver, D.; Hwang, H.S.; Davies, S.S.; Roach, D.E.; Duff, H.J.; Roden, D.M.; Wilde, A.A.M.; Knollmann, B.C. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 2009, 15, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Denegri, M.; Ruan, Y.; Avelino-Cruz, J.E.; Perissi, A.; Negri, S.; Napolitano, C.; Coetzee, W.A.; Boyden, P.A.; Priori, S.G. Short communication: Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ. Res. 2011, 109, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Tiso, N.; Salamon, M.; Bagattin, A.; Danieli, G.A.; Argenton, F.; Bortolussi, M. The binding of the RyR2 calcium channel to its gating protein FKBP12.6 is oppositely affected by ARVD2 and VTSIP mutations. Biochem. Biophys. Res. Commun. 2002, 299, 594–598. [Google Scholar] [CrossRef]
- Postma, A.V. Absence of Calsequestrin 2 Causes Severe Forms of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2002, 91, 21e–26e. [Google Scholar] [CrossRef]
- Song, L.; Alcalai, R.; Arad, M.; Wolf, C.M.; Toka, O.; Conner, D.A.; Berul, C.I.; Eldar, M.; Seidman, C.E.; Seidman, J.G. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2007, 117, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Bagnall, R.D.; Lam, L.; Ingles, J.; Turner, C.; Haan, E.; Davis, A.; Yang, P.C.; Clancy, C.E.; Sy, R.W.; et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016, 13, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in bedouin families from Israel. Am. J. Hum. Genet. 2002, 67, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Di Barletta, M.R.; Viatchenko-Karpinski, S.; Nori, A.; Memmi, M.; Terentyev, D.; Turcato, F.; Valle, G.; Rizzi, N.; Napolitano, C.; Gyorke, S.; et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 2006, 114, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Györke, I.; Hester, N.; Jones, L.R.; Györke, S. The Role of Calsequestrin, Triadin, and Junctin in Conferring Cardiac Ryanodine Receptor Responsiveness to Luminal Calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef]
- Roux-buisson, N.; Cacheux, M.; Fourest-lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Overgaard, M.T.; Sondergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ. Arrhythm. Electrophysiol. 2016, 9, e004161. [Google Scholar] [CrossRef] [PubMed]
- Devalla, H.D.; Gélinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.; Tadros, R.; Jiang, H.; Le Béchec, A.; et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef] [PubMed]
- El Refaey, M.M.; Mohler, P.J. Ankyrins and spectrins in cardiovascular biology and disease. Front. Physiol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Wilde, A.A.M. Catecholaminergic polymorphic ventricular tachycardia: From bench to bedside. Heart 2013, 99, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 2015, 344, 1173–1178. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Current | Effect | Function | Prevalence |
|---|---|---|---|---|---|
| GENES ENCODING ION CHANNEL SUBUNITS | |||||
| 1. Major LQTS-susceptibility genes | |||||
| KCNQ1 | KV7.1 (α-subunit of the voltage-dependent K+ channel) | ↓ IKs | loss-of-function | mediator of the slow component of the delayed rectifying potassium IKs current | ⋍40% (LQT1) |
| KCNH2 | KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) | ↓ IKr | loss-of-function | mediator of the rapid component of the delayed rectifying potassium IKr current | ⋍30% (LQT2) |
| SCN5A | NaV1.5 (α-subunit of the voltage-dependent Na+ channel) | ↑ INa | gain-of-function | mediator of the depolarizing inward sodium INa current | ⋍10% (LQT3) |
| 2. Rare LQTS-susceptibility genes | |||||
| By reducing outward currents | |||||
| KCNE1 | minK (β1-subunit of the voltage-dependent K+ channel) | ↓ IKs | loss-of-function | auxiliary protein modulator of KV7.1 and the IKs current | <1% |
| KCNE2 | MiRP1 (β2-subunit of the voltage-dependent K+ channel) | ↓ IKr | loss-of-function | auxiliary protein modulator of KV11.1 and the IKr current | <1% |
| KCNJ2 | Kir2.1 (inward rectifying K+ channel) | ↓ IK1 | loss-of-function, extra-cardiac manifestations | mediator of the inward rectifying potassium IK1 current | <1% (Andersen-Tawil syndrome, LQT7) |
| KCNJ5 | Kir3.4 (G protein-activated inward rectifying K+ channel 4) | ↓ IK,Ach | loss-of-function | mediator of the acetylcholine/adenosine-induced potassium IK,Ach current | <1% |
| By increasing inward currents | |||||
| SCN1B | β1-subunit of the voltage-dependent Na+ channel | ↑ INa | gain-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN4B | β4-subunit of the voltage-dependent Na+ channel | ↑ INa | gain-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| CACNA1C | CaV1.2 (α1C-subunit of the voltage-dependent L-type Ca2+ channel) | ↑ ICaL | gain-of-function, extra-cardiac manifestations | mediator of the inward calcium ICaL current | <1% (Timothy syndrome, LQT8) |
| GENES ENCODING AUXILIARY PROTEINS | |||||
| By reducing outward currents | |||||
| AKAP9 | A-kinase anchor protein-9 | ↓ IKs | disruption of KV7.1/PKA interaction | scaffolding protein assembling PKA and KV7.1 | <1% |
| By increasing inward currents | |||||
| ANK2 | ankyrin B | ↑ ICaL | disruption of Na+/K+ exchanger, Na+/Ca2+ exchanger/IP3 interaction | scaffolding protein assembling Na+/K+ exchanger, Na+/Ca2+ exchanger and IP3 receptor | <1% |
| CALM1 | calmodulin (CaM) | ↑ ICaL | disorder in CaV1.2 functioning | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 (and others) | <1% |
| CALM2 | calmodulin (CaM) | ↑ ICaL | disorder in CaV1.2 functioning | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 (and others) | <1% |
| CALM3 | calmodulin (CaM) | ↑ ICaL | disorder in CaV1.2 functioning | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 (and others) | <1% |
| SNTA1 | α1-syntrophin | ↑ INa | disruption of NaV1.5/NOS-PMCA4b complex interaction | scaffolding protein that associates NaV1.5 channels with the NOS-PMCA4b complex | <1% |
| TRDN | triadin | ↑ ICaL | reduction of ICaL inactivation | regulator of ryanodine receptors and CaV1.2 | <1% |
| Less established mechanisms | |||||
| CAV3 | caveolin-3 | ↑ INa?/↓ IK1? | changes in membrane expression of NaV1.5/Kir2.1 | scaffolding protein regulating ion channels in caveolae | <1% |
| TRPM4 | Transient receptor potential melastatin 4 | loss-of-function | regulator of conduction and cellular electrical activity which impact heart development | <1% | |
| RYR2 | ryanodine receptor 2 (RyR2) | not described | mediator of Ca2+ release from the SR | <1% | |
| Gene | Protein | Current | Effect | Function | Prevalence |
|---|---|---|---|---|---|
| GENES ENCODING ION CHANNEL SUBUNITS | |||||
| By increasing outward currents | |||||
| KCNH2 | KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) | ↑ IKr | gain-of-function | mediator of the rapid component of the delayed rectifying potassium IKr current | ⋍15% (SQT1) |
| KCNQ1 | KV7.1 (α-subunit of the voltage-dependent K+ channel) | ↑ IKs | gain-of-function | mediator of the slow component of the delayed rectifying potassium IKs current | <1% |
| KCNJ2 | Kir2.1 (inward rectifying K+ channel) | ↑ IK1 | gain-of-function | mediator of the inward rectifying potassium IK1 current | <1% |
| By decreasing inward currents | |||||
| CACNA1C | CaV1.2 (α1C-subunit of the voltage-dependent L-type Ca2+ channel) | ↓ ICaL | loss-of-function, combined phenotype of SQTS and BrS | mediator of the inward calcium ICaL current | <1% |
| CACNB2b | β2-subunit of the voltage-dependent L-type Ca2+ channel | ↓ ICaL | loss-of-function, combined phenotype of SQTS and BrS | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| Less established mechanisms | |||||
| CACNA2D1 | α2/δ-subunit of the voltage-dependent L-type Ca2+ channel | ↓ ICaL? | loss-of-function? | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| Gene | Protein | Current | Effect | Function | Prevalence |
|---|---|---|---|---|---|
| GENES ENCODING ION CHANNEL SUBUNITS | |||||
| 1. Major BrS-susceptibility genes | |||||
| SCN5A | NaV1.5 (α-subunit of the voltage-dependent Na+ channel) | ↓ INa | loss-of-function | mediator of the depolarizing inward sodium INa current | ⋍25% (BrS1) |
| 2. Rare BrS-susceptibility genes | |||||
| By decreasing inward currents | |||||
| SCN1B | β1-subunit of the voltage-dependent Na+ channel | ↓ INa | loss-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN2B | β2-subunit of the voltage-dependent Na+ channel | ↓ INa | loss-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN3B | β3-subunit of the voltage-dependent Na+ channel | ↓ INa | loss-of-function | auxiliary protein modulator of NaV1.5 and the INa current | <1% |
| SCN10A | NaV1.8 (α-subunit of the neuronal voltage-dependent Na+ channel) | ↓ INa | loss-of-function | mediator of the depolarizing phase of the neural AP, associated with pain perception | ⋍10%? |
| CACNA1C | CaV1.2 (α1C-subunit of the volatge-dependent L-type Ca2+ channel) | ↓ ICaL | loss-of-function, combined phenotype of BrS and SQTS | mediator of the inward calcium ICaL current | <1% |
| CACNB2b | β2-subunit of the voltage-dependent L-type Ca2+ channel | ↓ ICaL | loss-of-function, combined phenotype of BrS and SQTS | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| By increasing outward currents | |||||
| KCND3 | KV4.3 (α-subunit of the voltage-dependent K+ channel) | ↑ Ito | gain-of-function | mediator of the transient outward K+ Ito current | <1% |
| KCNE3 | minK-related peptide 2 (β-subunit of the voltage-dependent K+ channel) | ↑ Ito | gain-of-function | regulator of KV4.3 | <1% |
| KCNAB2 | β2-subunit of the voltage-dependent K+ channel | ↑ Ito | gain-of-function | interaction with KV4.3 | <1% |
| KCND2 | KV4.2 (voltage-dependent K+ channel) | ↑ Ito | gain-of-function | contributor to the transient outward K+ Ito current | <1% |
| KCNE5 | minK-related peptide 4 (β-subunit of the voltage-dependent K+ channel) | ↑ Ito | gain-of-function | inhibitor of the delayed rectifying KV7.1 channel and modulator of KV4.3 | <1% |
| KCNJ8 | Kir6.1 (inward-rectifier K+ channel, subunit of the ATP-sensitive K+ channel) | ↑ IK-ATP | gain-of-function | mediator of the IK-ATP currents | <1% |
| ABCC9 | SUR2 (sulfonylurea receptor, subunit of the ATP-sensitive K+ channel) | ↑ IK-ATP | gain-of-function | modulator of IK-ATP currents | <1% |
| KCNH2 | KV11.1/hERG (α-subunit of the voltage-dependent K+ channel) | ↑ IKr | gain-of-function | mediator of the rapid component of the delayed rectifying potassium IKr current | <1% |
| Less established mechanisms | |||||
| CACNA2D1 | α2/δ subunit of the volatge-dependent L-type Ca2+ channel | ↓ ICaL? | loss-of-function?, combined phenotype of SQTS and BrS | auxiliary protein modulator of CaV1.2 and the ICaL current | <1% |
| HCN4 | hyperpolarization-activated, cyclic nucleotide-gated ion channel 4 | ↓ If? | loss-of-function? | mediator of the pacemaker current, If | <1% |
| TRPM4 | Transient receptor potential melastatin 4 | loss-of-function/gain-of-function | regulator of conduction and cellular electrical activity which impact heart development | <1% | |
| GENES ENCONDING AUXILIARY PROTEINS | |||||
| FGF12 | fibroblast growth factor 12 | ↓ INa | interaction with NaV1.5 trafficking | modulator of Nav1.5 and the INa current | <1% |
| GPD1L | glycerol-3-phosphate dehydrogenase 1-like | ↓ INa | interaction with NaV1.5 trafficking | modulator of Na1.5 and the INa current | <1% |
| SLMAP | sarcolemma associated protein (striatin-interacting phosphatase and kinase complex) | ↓ INa | interaction with NaV1.5 trafficking | present in the T-tubules, regulator of excitation-contraction coupling | <1% |
| PKP2 | plakophillin-2 | ↓ INa | changes in NaV1.5 expression in intercalated disc | binds to and modulates NaV1.5 and the INa current | <1% |
| SEMA3A | semaphorin-3A | ↑ Ito | loss-of-function | inhibitor of the KV4.3 channel | <1% |
| Less established mechanisms | |||||
| RANGRF | MOG1 (multicopy suppressor of Gsp1) | ↓ INa? | interaction with NaV1.5 trafficking | involved in nuclear protein import—regulates cell surface location of NaV1.5 | <1% |
| HEY2 | CHF1 (cardiovascular helix-loop-helix factor 1) | ↑ Ito? | interaction with KCNIP2 | transcriptional regulator of cardiac electrical function | <1% |
| Gene | Protein | Effect | Function | Prevalence |
|---|---|---|---|---|
| GENES ENCODING ION CHANNELS AND AUXILIARY PROTEINS | ||||
| 1. Major CPVT-susceptibility genes | ||||
| RYR2 | ryanodine receptor 2 (RyR2) | cytoplasmic Ca2+ overload, due to Ca2+ leak from the SR | mediator of the release of stored Ca2+ ions from the SR | ⋍50–60% (CPVT1) |
| CASQ2 | calsequestrin 2 | decreased Ca2+ content in the SR and abnormal Ca2+ regulation | Ca2+ storage protein, controls Ca2+ release from the SR | ⋍5% |
| 2. Rare CPVT-susceptibility genes | ||||
| TRDN | triadin | cytoplasmic Ca2+ overload, due to Ca2+ leak from the SR | regulator of ryanodine receptors, controls the Ca2+ release from the SR | <1% |
| CALM1 | calmodulin (CaM) | Ca2+ leak from the SR due to loss of interaction CaM-RyR2 | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 or RyR2 (and others) | <1% |
| CALM2 | calmodulin (CaM) | reduction in Ca2+-binding affinity in the CaM C-domain | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 or RyR2 (and others) | <1% |
| CALM3 | calmodulin (CaM) | reduction in Ca2+-binding affinity in the CaM C-domain and leak from the SR | essential Ca2+ sensor, signal-transducing protein modulator of CaV1.2 or RyR2 (and others) | <1% |
| TECLR | trans-2,3-enoyl-CoA reductase- like | decreased Ca2+ content in the SR and abnormal Ca2+ regulation | participates in the synthesis of fatty acids | <1% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Elias, A.; Benito, B. Ion Channel Disorders and Sudden Cardiac Death. Int. J. Mol. Sci. 2018, 19, 692. https://doi.org/10.3390/ijms19030692
Garcia-Elias A, Benito B. Ion Channel Disorders and Sudden Cardiac Death. International Journal of Molecular Sciences. 2018; 19(3):692. https://doi.org/10.3390/ijms19030692
Chicago/Turabian StyleGarcia-Elias, Anna, and Begoña Benito. 2018. "Ion Channel Disorders and Sudden Cardiac Death" International Journal of Molecular Sciences 19, no. 3: 692. https://doi.org/10.3390/ijms19030692
APA StyleGarcia-Elias, A., & Benito, B. (2018). Ion Channel Disorders and Sudden Cardiac Death. International Journal of Molecular Sciences, 19(3), 692. https://doi.org/10.3390/ijms19030692