Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis


Abstract
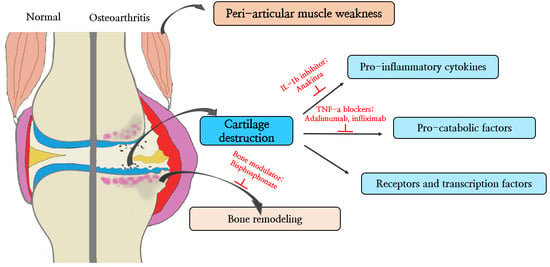
{kind=link}
1. Introduction
2. Molecular Pathology of Cartilage Destruction
2.1. Pro-Inflammatory Cytokines
2.2. Pro-Catabolic Factors
2.3. Transcription Factors
2.4. Inherent Changes in Chondrocytes: Senescence, Apoptosis, Autophagy
3. Molecular Pathology of Bone and Peri-Articular Muscle
4. Novel Therapeutics Based on the Molecular Pathogenesis of OA
4.1. Anti-Inflammatory and Cytokine Blocker
4.1.1. Anti TNF-α Therapies
4.1.2. IL-1β Signalling Inhibitors
4.1.3. NO Inhibitors
4.2. Bone Modulators
4.2.1. Bisphosphonates
4.2.2. Strontium Ranelate
4.2.3. NGF Inhibitors
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iannone, F.; Lapadula, G. The pathophysiology of osteoarthritis. Aging Clin. Exp. Res. 2003, 15, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Mortellaro, C.M. Pathophysiology of osteoarthritis. Vet. Res. Commun. 2003, 27 (Suppl. S1), 75–78. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T. Clinical practice. Osteoarthritis of the knee. N. Engl. J. Med. 2006, 354, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Krasnokutsky, S.; Samuels, J.; Abramson, S.B. Osteoarthritis in 2007. Bull. NYU Hosp. Jt. Dis. 2007, 65, 222–228. [Google Scholar] [PubMed]
- Musumeci, G.; Loreto, C.; Carnazza, M.L.; Martinez, G. Characterization of apoptosis in articular cartilage derived from the knee joints of patients with osteoarthritis. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E.; Brandt, K.; Hawker, G.; Peeva, E.; Schreyer, E.; Tsuji, W.; Hochberg, M.C. OARSI-FDA initiative: Defining the disease state of osteoarthritis. Osteoarthr. Cartil. 2011, 19, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Ellman, M.B.; Yan, D.; Kroin, J.S.; Cole, B.J.; van Wijnen, A.J.; Im, H.J. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene 2013, 527, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Im, H.J.; Muddasani, P.; Natarajan, V.; Schmid, T.M.; Block, J.A.; Davis, F.; van Wijnen, A.J.; Loeser, R.F. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cδ pathways in human adult articular chondrocytes. J. Biol. Chem. 2007, 282, 11110–11121. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.; Nam, J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. BioMed Res. Int. 2013, 2013, 284873. [Google Scholar] [CrossRef] [PubMed]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Van der Kraan, P.M.; van den Berg, W.B. Osteophytes: Relevance and biology. Osteoarthr. Cartil. 2007, 15, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.; Englund, M.; Struglics, A.; Lohmander, L.S. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthr. Cartil. 2015, 23, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Vergunst, C.E.; van de Sande, M.G.; Lebre, M.C.; Tak, P.P. The role of chemokines in rheumatoid arthritis and osteoarthritis. Scand. J. Rheumatol. 2005, 34, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Sohn, D.H.; Sokolove, J.; Sharpe, O.; Erhart, J.C.; Chandra, P.E.; Lahey, L.J.; Lindstrom, T.M.; Hwang, I.; Boyer, K.A.; Andriacchi, T.P.; et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res. Ther. 2012, 14, R7. [Google Scholar] [CrossRef] [PubMed]
- Dayer, J.M. The process of identifying and understanding cytokines: From basic studies to treating rheumatic diseases. Best Pract. Res. Clin. Rheumatol. 2004, 18, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, P.S.; Varadarajan, U.; Sheldon, R.; Fortier, E.; LaVallie, E.R.; Morris, E.A.; Yaworsky, P.J.; Majumdar, M.K. Involvement of protein kinase Czeta in interleukin-1beta induction of ADAMTS-4 and type 2 nitric oxide synthase via NF-kappaB signaling in primary human osteoarthritic chondrocytes. Arthritis Rheum. 2007, 56, 4074–4083. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.M.; Price, J.S.; Chambers, M.G.; Visco, D.M.; Poole, A.R.; Mason, R.M. Gene deletion of either interleukin-1beta, interleukin-1beta-converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy. Arthritis Rheum. 2003, 48, 3452–3463. [Google Scholar] [PubMed]
- Kobayashi, M.; Squires, G.R.; Mousa, A.; Tanzer, M.; Zukor, D.J.; Antoniou, J.; Feige, U.; Poole, A.R. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005, 52, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Saklatvala, J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 1986, 322, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Peeters-Joris, C.; Vaes, G. Modulation by interleukin 1 and tumor necrosis factor alpha of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim. Biophys. Acta 1990, 1052, 366–378. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Moilanen, T.; Hamalainen, M.; Moilanen, E. Effects of TNFalpha-antagonists on nitric oxide production in human cartilage. Osteoarthr. Cartil. 2002, 10, 327–332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goldring, M.B. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000, 43, 1916–1926. [Google Scholar] [CrossRef]
- Mueller, M.B.; Tuan, R.S. Anabolic/Catabolic balance in pathogenesis of osteoarthritis: Identifying molecular targets. PM R 2011, 3 (Suppl. S1), S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Taskiran, D.; Stefanovic-Racic, M.; Georgescu, H.; Evans, C. Nitric oxide mediates suppression of cartilage proteoglycan synthesis by interleukin-1. Biochem. Biophys. Res. Commun. 1994, 200, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sahebjam, S.; Khokha, R.; Mort, J.S. Increased collagen and aggrecan degradation with age in the joints of Timp3(−/−) mice. Arthritis Rheum. 2007, 56, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Sylvester, J.; Ahmad, M.; Zafarullah, M. Adaptor proteins and Ras synergistically regulate IL-1-induced ADAMTS-4 expression in human chondrocytes. J. Immunol. 2009, 182, 5081–5087. [Google Scholar] [CrossRef] [PubMed]
- East, C.J.; Stanton, H.; Golub, S.B.; Rogerson, F.M.; Fosang, A.J. ADAMTS-5 deficiency does not block aggrecanolysis at preferred cleavage sites in the chondroitin sulfate-rich region of aggrecan. J. Biol. Chem. 2007, 282, 8632–8640. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Saito, T.; Sugita, S.; Hikata, T.; Kobayashi, H.; Fukai, A.; Taniguchi, Y.; Hirata, M.; Akiyama, H.; Chung, U.I.; et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 2013, 110, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Hosaka, Y.; Okada, K.; Mori, D.; Yano, F.; Kobayashi, H.; Taniguchi, Y.; Mori, Y.; Okuma, T.; Chang, S.H.; et al. Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl. Acad. Sci. USA 2015, 112, 3080–3085. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, J.; Mirando, A.J.; Wang, C.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci. Signal. 2015, 8, ra71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.J.; Luo, W.; Lei, G.H. Role of HIF-1alpha and HIF-2alpha in osteoarthritis. Jt. Bone Spine 2015, 82, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Pfander, D.; Kobayashi, T.; Knight, M.C.; Zelzer, E.; Chan, D.A.; Olsen, B.R.; Giaccia, A.J.; Johnson, R.S.; Haase, V.H.; Schipani, E. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development 2004, 131, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Schipani, E.; Ryan, H.E.; Didrickson, S.; Kobayashi, T.; Knight, M.; Johnson, R.S. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001, 15, 2865–2876. [Google Scholar] [PubMed]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Rigoglou, S.; Papavassiliou, A.G. The NF-kappaB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 2013, 45, 2580–2584. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.X.; Lin, L.; Wang, H.J.; Wei, X.L.; Fu, X.; Zhang, J.Y.; Yu, C.L. Suppression of early experimental osteoarthritis by in vivo delivery of the adenoviral vector-mediated NF-kappaBp65-specific siRNA. Osteoarthr. Cartil. 2008, 16, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Lauder, S.; Wainwright, S.; Amos, N.; Evans, A.; Hughes, C.; Feldmann, M.; Caterson, B. Adenoviral gene transfer of the endogenous inhibitor IκBα into human osteoarthritis synovial fibroblasts demonstrates that several matrix metalloproteinases and aggrecanases are nuclear factor-κB-dependent. J. Rheumatol. 2007, 34, 523–533. [Google Scholar] [PubMed]
- Amos, N.; Lauder, S.; Evans, A.; Feldmann, M.; Bondeson, J. Adenoviral gene transfer into osteoarthritis synovial cells using the endogenous inhibitor IkappaBalpha reveals that most, but not all, inflammatory and destructive mediators are NFkappaB dependent. Rheumatology 2006, 45, 1201–1209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.H.; Jeon, J.; Shin, M.; Won, Y.; Lee, M.; Kwak, J.S.; Lee, G.; Rhee, J.; Ryu, J.H.; Chun, C.H.; et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 2014, 156, 730–743. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.D., Jr.; Loeser, R.F. Increased oxidative stress with aging reduces chondrocyte survival: Correlation with intracellular glutathione levels. Arthritis Rheum. 2003, 48, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Blake, S.; Song, X.Y.; Lark, M.; Loeser, R.F. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. Ther. 2008, 10, R23. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Heraud, F.; Heraud, A.; Harmand, M.F. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000, 59, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Suh, D.I.; Song, Y.W. Relationship between chondrocyte apoptosis and matrix depletion in human articular cartilage. J. Rheumatol. 2001, 28, 2038–2045. [Google Scholar] [PubMed]
- Pelletier, J.P.; Fernandes, J.C.; Jovanovic, D.V.; Reboul, P.; Martel-Pelletier, J. Chondrocyte death in experimental osteoarthritis is mediated by MEK 1/2 and p38 pathways: Role of cyclooxygenase-2 and inducible nitric oxide synthase. J. Rheumatol. 2001, 28, 2509–2519. [Google Scholar] [PubMed]
- Loening, A.M.; James, I.E.; Levenston, M.E.; Badger, A.M.; Frank, E.H.; Kurz, B.; Nuttall, M.E.; Hung, H.H.; Blake, S.M.; Grodzinsky, A.J.; et al. Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch. Biochem. Biophys. 2000, 381, 205–212. [Google Scholar] [CrossRef] [PubMed]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006, 54, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012, 64, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.A.; Adams, C.S.; Garcia, J.A.; Shapiro, I.M.; Srinivas, V. Regulation of autophagy in human and murine cartilage: Hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum. 2009, 60, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Wei, L.; Li, W.; Yang, W.; Cai, L.; Qian, Z.; Wu, S. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J. Pharmacol. Sci. 2017, 134, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, L.; Zhang, Y.; Cui, L.; Shen, G. Intra-articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF-2alpha. J. Orthop. Res. 2015, 33, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R. Role of bone in osteoarthritis pathogenesis. Med. Clin. N. Am. 2009, 93, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Bae, W.C.; Shieu, W.; Lewis, C.W.; Bugbee, W.D.; Sah, R.L. Increased hydraulic conductance of human articular cartilage and subchondral bone plate with progression of osteoarthritis. Arthritis Rheum. 2008, 58, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Day, J.S.; Ding, M.; van der Linden, J.C.; Hvid, I.; Sumner, D.R.; Weinans, H. A decreased subchondral trabecular bone tissue elastic modulus is associated with pre-arthritic cartilage damage. J. Orthop. Res. 2001, 19, 914–918. [Google Scholar] [CrossRef]
- Felson, D.T.; Gale, D.R.; Elon Gale, M.; Niu, J.; Hunter, D.J.; Goggins, J.; Lavalley, M.P. Osteophytes and progression of knee osteoarthritis. Rheumatology 2005, 44, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.L.; Meng, H.Y.; Wang, Y.C.; Peng, J.; Guo, Q.Y.; Wang, A.Y.; Lu, S.B. Bone-cartilage interface crosstalk in osteoarthritis: Potential pathways and future therapeutic strategies. Osteoarthr. Cartil. 2014, 22, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wluka, A.E.; Pelletier, J.P.; Martel-Pelletier, J.; Abram, F.; Ding, C.; Cicuttini, F.M. Meniscal extrusion predicts increases in subchondral bone marrow lesions and bone cysts and expansion of subchondral bone in osteoarthritic knees. Rheumatology 2010, 49, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Bai, L. Recent advances in the understanding of molecular mechanisms of cartilage degeneration, synovitis and subchondral bone changes in osteoarthritis. Connect. Tissue Res. 2016, 57, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Lories, R.J.; Peeters, J.; Bakker, A.; Tylzanowski, P.; Derese, I.; Schrooten, J.; Thomas, J.T.; Luyten, F.P. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007, 56, 4095–4103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Schivo, S.; Huang, X.; Leijten, J.; Karperien, M.; Post, J.N. Nitric Oxide Mediates Crosstalk between Interleukin 1beta and WNT Signaling in Primary Human Chondrocytes by Reducing DKK1 and FRZB Expression. Int. J. Mol. Sci. 2017, 18, 2491. [Google Scholar] [CrossRef] [PubMed]
- Bennell, K.L.; Hunt, M.A.; Wrigley, T.V.; Lim, B.W.; Hinman, R.S. Role of muscle in the genesis and management of knee osteoarthritis. Rheum. Dis. Clin. N. Am. 2008, 34, 731–754. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.V.; Scott, D.L.; Rees, J.; Newham, D.J. Sensorimotor changes and functional performance in patients with knee osteoarthritis. Ann. Rheum. Dis. 1997, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Liikavainio, T.; Lyytinen, T.; Tyrvainen, E.; Sipila, S.; Arokoski, J.P. Physical function and properties of quadriceps femoris muscle in men with knee osteoarthritis. Arch. Phys. Med. Rehabil. 2008, 89, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.H.; Zeni, J.A.; Snyder-Mackler, L. Muscle impairments in patients with knee osteoarthritis. Sports Health 2012, 4, 284–292. [Google Scholar] [CrossRef] [PubMed]
- De Ceuninck, F.; Fradin, A.; Pastoureau, P. Bearing arms against osteoarthritis and sarcopenia: When cartilage and skeletal muscle find common interest in talking together. Drug Discov. Today 2014, 19, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Rehan Youssef, A.; Longino, D.; Seerattan, R.; Leonard, T.; Herzog, W. Muscle weakness causes joint degeneration in rabbits. Osteoarthr. Cartil. 2009, 17, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Isaac, C.; Wright, A.; Usas, A.; Li, H.; Tang, Y.; Mu, X.; Greco, N.; Dong, Q.; Vo, N.; Kang, J.; et al. Dystrophin and utrophin “double knockout” dystrophic mice exhibit a spectrum of degenerative musculoskeletal abnormalities. J. Orthop. Res. 2013, 31, 343–349. [Google Scholar] [CrossRef] [PubMed]
- van der Poel, C.; Levinger, P.; Tonkin, B.A.; Levinger, I.; Walsh, N.C. Impaired muscle function in a mouse surgical model of post-traumatic osteoarthritis. Osteoarthr. Cartil. 2016, 24, 1047–1053. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peake, J.; Della Gatta, P.; Cameron-Smith, D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1485–R1495. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P. Molecular regulation of skeletal muscle mass. Clin. Exp. Pharmacol. Physiol. 2010, 37, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Fisher, P.B. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2006, 236, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Levinger, I.; Levinger, P.; Trenerry, M.K.; Feller, J.A.; Bartlett, J.R.; Bergman, N.; McKenna, M.J.; Cameron-Smith, D. Increased inflammatory cytokine expression in the vastus lateralis of patients with knee osteoarthritis. Arthritis Rheum. 2011, 63, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Rau, R. Adalimumab (a fully human anti-tumour necrosis factor alpha monoclonal antibody) in the treatment of active rheumatoid arthritis: The initial results of five trials. Ann. Rheum. Dis. 2002, 61 (Suppl. S2), ii70–ii73. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, G.; Wittoek, R.; Vander Cruyssen, B.; Elewaut, D. Tumour necrosis factor blockade for the treatment of erosive osteoarthritis of the interphalangeal finger joints: A double blind, randomised trial on structure modification. Ann. Rheum. Dis. 2012, 71, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, X.; Ravaud, P.; Maheu, E.; Baron, G.; Rialland, A.; Vergnaud, P.; Roux, C.; Maugars, Y.; Mulleman, D.; Lukas, C.; et al. Adalimumab in patients with hand osteoarthritis refractory to analgesics and NSAIDs: A randomised, multicentre, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2015, 74, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Guler-Yuksel, M.; Allaart, C.F.; Watt, I.; Goekoop-Ruiterman, Y.P.; de Vries-Bouwstra, J.K.; van Schaardenburg, D.; van Krugten, M.V.; Dijkmans, B.A.; Huizinga, T.W.; Lems, W.F.; et al. Treatment with TNF-alpha inhibitor infliximab might reduce hand osteoarthritis in patients with rheumatoid arthritis. Osteoarthr. Cartil. 2010, 18, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Efficacy and safety of adalimumab by intra-articular injection for moderate to severe knee osteoarthritis: An open-label randomized controlled trial. J. Int. Med. Res. 2018, 46, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Haywood, L.; McWilliams, D.F.; Pearson, C.I.; Gill, S.E.; Ganesan, A.; Wilson, D.; Walsh, D.A. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum. 2003, 48, 2173–2177. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Osteoarthritis of the Knee, Inflammation, and the Effect of Adalimumab (OKINADA) (OKINADA). Canadian Research & Education in Arthritis. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT02471118 (accessed on 11 April 2017).
- Martel-Pelletier, J.; Pelletier, J.P. Effects of diacerein at the molecular level in the osteoarthritis disease process. Ther. Adv. Musculoskelet. Dis. 2010, 2, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Dougados, M.; Nguyen, M.; Berdah, L.; Mazieres, B.; Vignon, E.; Lequesne, M.; Group, E.I.S. Evaluation of the structure-modifying effects of diacerein in hip osteoarthritis: ECHODIAH, a three-year, placebo-controlled trial. Evaluation of the Chondromodulating Effect of Diacerein in OA of the Hip. Arthritis Rheum. 2001, 44, 2539–2547. [Google Scholar] [CrossRef]
- Fidelix, T.S.; Macedo, C.R.; Maxwell, L.J.; Fernandes Moca Trevisani, V. Diacerein for osteoarthritis. Cochrane Database Syst. Rev. 2014, CD005117. [Google Scholar] [CrossRef] [PubMed]
- Calich, A.L.; Domiciano, D.S.; Fuller, R. Osteoarthritis: Can anti-cytokine therapy play a role in treatment? Clin. Rheumatol. 2010, 29, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Bacconnier, L.; Jorgensen, C.; Fabre, S. Erosive osteoarthritis of the hand: Clinical experience with anakinra. Ann. Rheum. Dis. 2009, 68, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009, 61, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Sorrenti, V.; Di Giacomo, C.; Romeo, G.; Siracusa, M.A. Progress in the development of selective nitric oxide synthase (NOS) inhibitors. Curr. Pharm. Des. 2002, 8, 177–200. [Google Scholar] [CrossRef] [PubMed]
- More, A.S.; Kumari, R.R.; Gupta, G.; Lingaraju, M.C.; Balaganur, V.; Pathak, N.N.; Kumar, D.; Kumar, D.; Sharma, A.K.; Tandan, S.K. Effect of iNOS inhibitor S-methylisothiourea in monosodium iodoacetate-induced osteoathritic pain: Implication for osteoarthritis therapy. Pharmacol. Biochem. Behav. 2013, 103, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Hellio le Graverand, M.P.; Clemmer, R.S.; Redifer, P.; Brunell, R.M.; Hayes, C.W.; Brandt, K.D.; Abramson, S.B.; Manning, P.T.; Miller, C.G.; Vignon, E. A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010), in patients with symptomatic osteoarthritis of the knee. Ann. Rheum. Dis. 2013, 72, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Smith, T.O.; Hing, C.B.; Sofat, N. Are bisphosphonates effective in the treatment of osteoarthritis pain? A meta-analysis and systematic review. PLoS ONE 2013, 8, e72714. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, K.; Lan, Y.; Zhang, N.; Jiang, R.; Hu, J. Alendronate protects against articular cartilage erosion by inhibiting subchondral bone loss in ovariectomized rats. Bone 2013, 53, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Strassle, B.W.; Mark, L.; Leventhal, L.; Piesla, M.J.; Jian Li, X.; Kennedy, J.D.; Glasson, S.S.; Whiteside, G.T. Inhibition of osteoclasts prevents cartilage loss and pain in a rat model of degenerative joint disease. Osteoarthr. Cartil. 2010, 18, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Bingham, C.O., III; Buckland-Wright, J.C.; Garnero, P.; Cohen, S.B.; Dougados, M.; Adami, S.; Clauw, D.J.; Spector, T.D.; Pelletier, J.P.; Raynauld, J.P.; et al. Risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or slow radiographic progression in patients with medial compartment osteoarthritis of the knee: Results of the two-year multinational knee osteoarthritis structural arthritis study. Arthritis Rheum. 2006, 54, 3494–3507. [Google Scholar] [PubMed]
- Laslett, L.L.; Dore, D.A.; Quinn, S.J.; Boon, P.; Ryan, E.; Winzenberg, T.M.; Jones, G. Zoledronic acid reduces knee pain and bone marrow lesions over 1 year: A randomised controlled trial. Ann. Rheum. Dis. 2012, 71, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.L.; Zhao, L.R.; Wang, P.M. Bisphosphonates therapy for osteoarthritis: A meta-analysis of randomized controlled trials. SpringerPlus 2016, 5, 1704. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Valenti, M.T.; Mottes, M.; Biotti, A.; Perduca, M.; Pisani, A.; Bovi, M.; Deiana, M.; Cheri, S.; Dalle Carbonare, L. Clodronate as a Therapeutic Strategy against Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 2696. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Adami, S.; Fracassi, E.; Viapiana, O.; Orsolini, G.; Povino, M.R.; Idolazzi, L.; Gatti, D. Effects of intra-articular clodronate in the treatment of knee osteoarthritis: Results of a double-blind, randomized placebo-controlled trial. Rheumatol. Int. 2015, 35, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Saviola, G.; Abdi-Ali, L.; Povino, M.R.; Campostrini, L.; Sacco, S.; Carbonare, L.D. Intramuscular clodronate in erosive osteoarthritis of the hand is effective on pain and reduces serum COMP: A randomized pilot trial—The ER.O.D.E. study (ERosive Osteoarthritis and Disodium-clodronate Evaluation). Clin. Rheumatol. 2017, 36, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Meunier, P.J.; Roux, C.; Seeman, E.; Ortolani, S.; Badurski, J.E.; Spector, T.D.; Cannata, J.; Balogh, A.; Lemmel, E.M.; Pors-Nielsen, S.; et al. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N. Engl. J. Med. 2004, 350, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.D. Clinical effects of strontium ranelate in women with postmenopausal osteoporosis. Osteoporos. Int. 2005, 16 (Suppl. S1), S16–S19. [Google Scholar] [CrossRef] [PubMed]
- Reginster, J.Y.; Badurski, J.; Bellamy, N.; Bensen, W.; Chapurlat, R.; Chevalier, X.; Christiansen, C.; Genant, H.; Navarro, F.; Nasonov, E.; et al. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: Results of a double-blind, randomised placebo-controlled trial. Ann. Rheum. Dis. 2013, 72, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.G.; Ding, H.F.; Mao, Y.Q.; Liu, M.; Yu, B.; Zhao, X.; Wang, X.Q.; Li, Y.; Liu, G.W.; Nie, S.B.; et al. Strontium ranelate reduces cartilage degeneration and subchondral bone remodeling in rat osteoarthritis model. Acta Pharmacol. Sin. 2013, 34, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Roubille, C.; Raynauld, J.P.; Abram, F.; Dorais, M.; Delorme, P.; Martel-Pelletier, J. Disease-modifying effect of strontium ranelate in a subset of patients from the Phase III knee osteoarthritis study SEKOIA using quantitative MRI: Reduction in bone marrow lesions protects against cartilage loss. Ann. Rheum. Dis. 2015, 74, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, T.J.; Ekman, E.F.; Spierings, E.L.; Greenberg, H.S.; Smith, M.D.; Brown, M.T.; West, C.R.; Verburg, K.M. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann. Rheum. Dis. 2015, 74, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C.; Tive, L.A.; Abramson, S.B.; Vignon, E.; Verburg, K.M.; West, C.R.; Smith, M.D.; Hungerford, D.S. When Is Osteonecrosis Not Osteonecrosis?: Adjudication of Reported Serious Adverse Joint Events in the Tanezumab Clinical Development Program. Arthritis Rheumatol. 2016, 68, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. A Study to Determine the Safety and the Efficacy of Fasinumab Compared to Placebo and Naproxen for Treatment of Adults with Pain From Osteoarthritis of the Knee or Hip (FACT OA1). Regeneron Pharmaceuticals. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03161093 (accessed on 29 August 2017).
- Clinicaltrials.gov. Long-Term Safety and Efficacy Study of Fasinumab in Patients with Pain Due to Osteoarthritis (OA) of the Knee or Hip (FACT LTS & OA). Regeneron Pharmaceuticals. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02683239 (accessed on 18 May 2017).
- Clinicaltrials.gov. Study to Determine the Safety and the Efficacy of Fasinumab Compared to Placebo and Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) for Treatment of Adults with Pain from Osteoarthritis of the Knee or Hip (FACT OA2). Regeneron Pharmaceuticals. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03304379 (accessed on 6 December 2017).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-R.; Yoo, J.J.; Kim, H.A. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 674. https://doi.org/10.3390/ijms19030674
Kim J-R, Yoo JJ, Kim HA. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. International Journal of Molecular Sciences. 2018; 19(3):674. https://doi.org/10.3390/ijms19030674
Chicago/Turabian StyleKim, Ju-Ryoung, Jong Jin Yoo, and Hyun Ah Kim. 2018. "Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis" International Journal of Molecular Sciences 19, no. 3: 674. https://doi.org/10.3390/ijms19030674
APA StyleKim, J.-R., Yoo, J. J., & Kim, H. A. (2018). Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. International Journal of Molecular Sciences, 19(3), 674. https://doi.org/10.3390/ijms19030674