Mesenchymal Stem Cell Protection of Neurons against Glutamate Excitotoxicity Involves Reduction of NMDA-Triggered Calcium Responses and Surface GluR1, and Is Partly Mediated by TNF


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
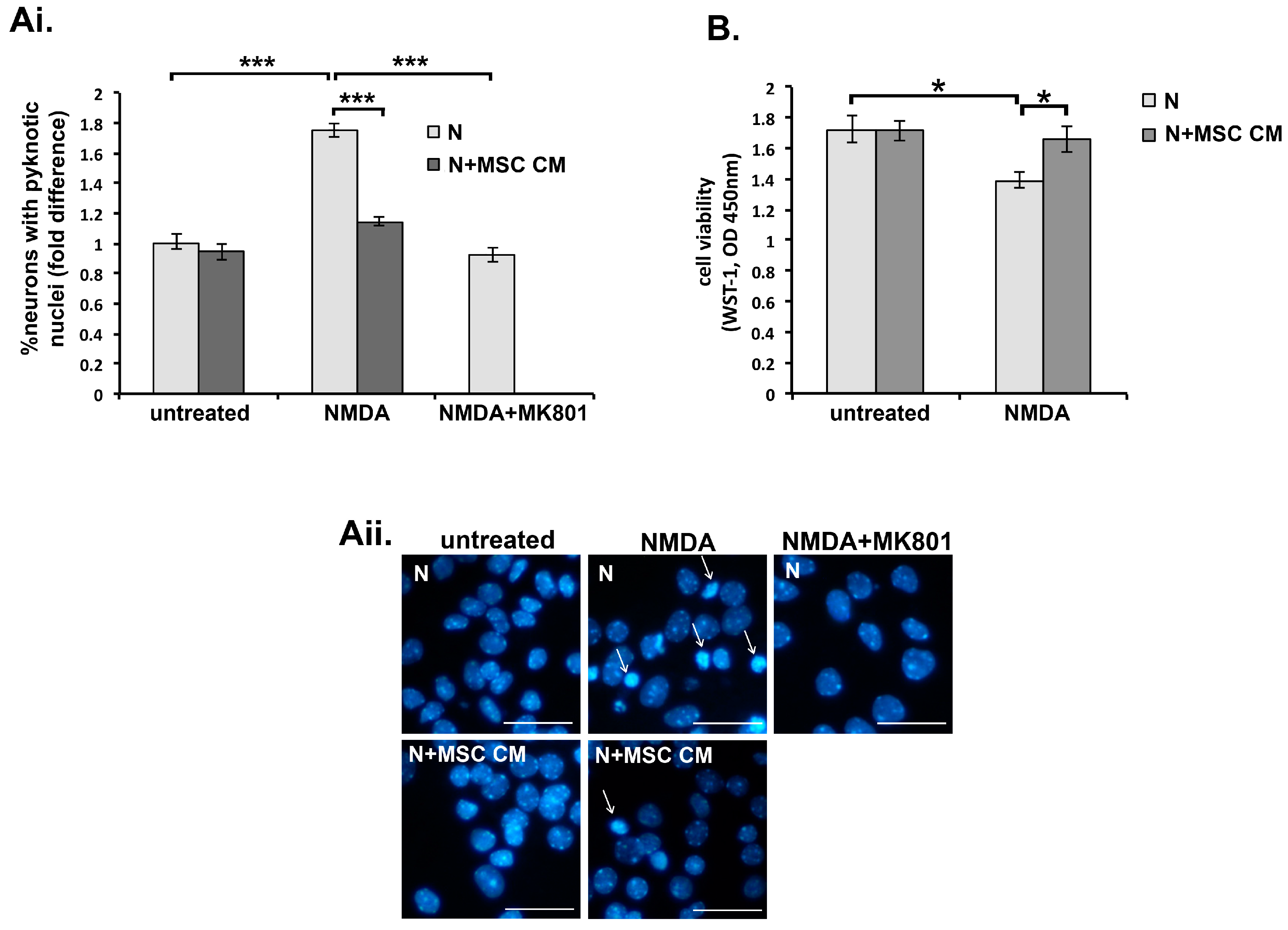
2.1. MSC Conditioned Medium Protects Isolated Mouse Cortical Neurons against Glutamate Excitotoxicity
2.2. Tumor Necrosis Factor Is Produced by MSC and Contributes to MSC CM-Mediated Neuroprotection
2.3. MSC Secreted Factors Do Not Induce Differential Survival of Neurons or Expansion of Non-Neuronal Cells in Long-Term Cultures
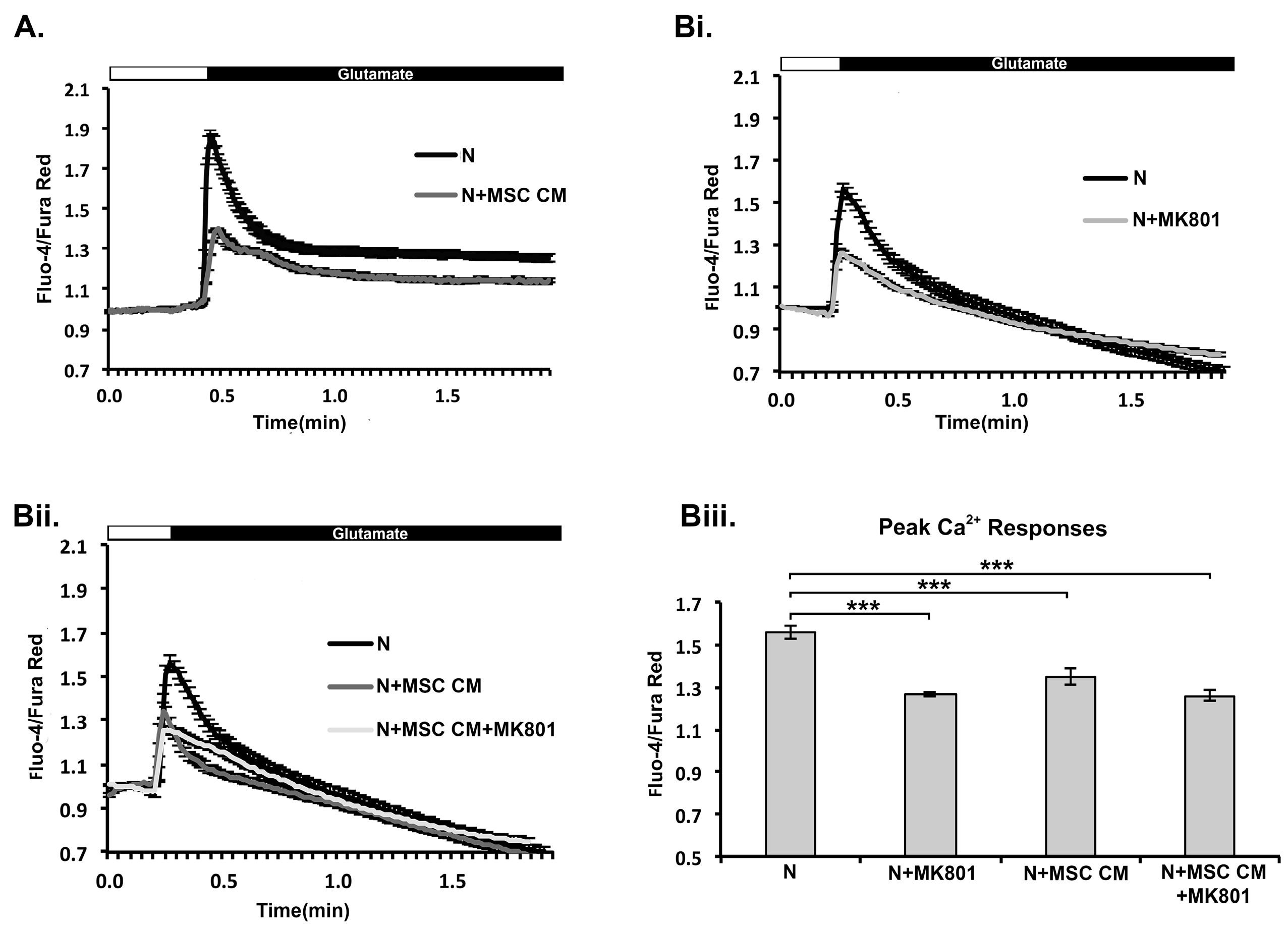
2.4. MSC CM Prevents Increase in Ca2+ Levels in Mouse Cortical Neurons in Response to Glutamate Receptor Stimulation
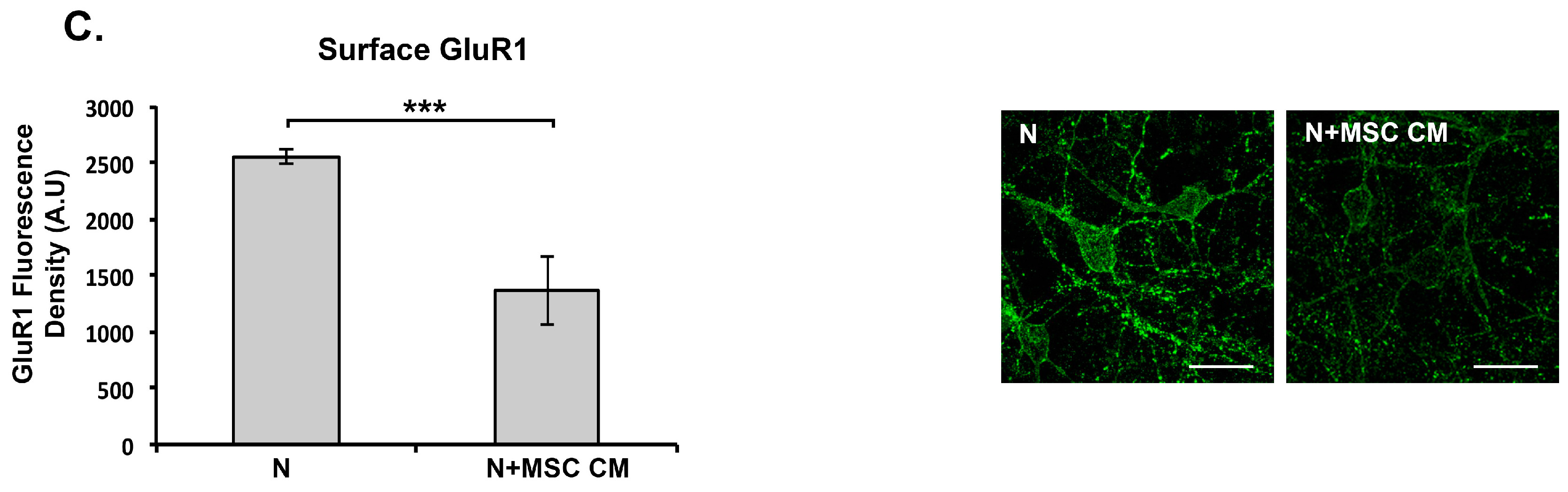
2.5. MSC CM Reduces Cell Surface Expression of the GluR1 Subunit of the AMPAR on Cortical Neurons
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture and Experimental Treatments
4.3. TNF Bioactivity and Cell Death Assays
4.4. Immunocytochemistry
4.5. Calcium Imaging
4.6. Real-Time Quantitative RT-PCR
4.7. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPAR | AMPA receptors |
| Ara C | Arabinose C |
| BBB | Blood brain barrier |
| Ca2+ | Calcium |
| CNS | Central nervous system |
| DAPI | 4′-6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| EAE | Experimental autoimmune encephalomyelitis |
| FBS | Fetal bovine serum |
| FGF-II | Fibroblast growth factor-II |
| GluR1 | AMPAR subunit |
| hgDMEM | High glucose DMEM |
| MSC | Mesenchymal stem cells |
| MSC CM | Mesenchymal stem cells conditioned medium |
| NeuN | Neuronal nuclei |
| NPC | Neural precursor cells |
| NG2 | Neural/glial antigen 2 |
| NMDA | N-Methyl-d-aspartic acid |
| NMDAR | NMDA receptor |
| RGC | Retinal ganglion cells |
| TNF | Tumor necrosis factor |
References
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.; Friedenstein, A.J. Stromal stem cells: Marrow-derived osteogenic precursors. Ciba Found. Symp. 1988, 136, 42–60. [Google Scholar] [PubMed]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Latsinik, N.V.; Panasyuk, A.F.; Keiliss-Borok, I.V. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation 1974, 17, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Laroni, A.; Freedman, M.S. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet. Neurol. 2011, 10, 649–656. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The msc: An injury drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Chen, X.G.; Wang, L.; Gautam, S.C.; Xu, Y.X.; Katakowski, M.; Zhang, L.J.; Lu, M.; Janakiraman, N.; et al. Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology 2002, 59, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Katakowski, M.; Chen, X.; Wang, L.; Lu, D.; Lu, M.; Gautam, S.C.; Chopp, M. Intravenous bone marrow stromal cell therapy reduces apoptosis and promotes endogenous cell proliferation after stroke in female rat. J. Neurosci. Res. 2003, 73, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Ohtaki, H.; Ylostalo, J.H.; Foraker, J.E.; Robinson, A.P.; Reger, R.L.; Shioda, S.; Prockop, D.J. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc. Natl. Acad. Sci. USA 2008, 105, 14638–14643. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Radtke, C.; Honmou, O.; Kocsis, J.D. Remyelination of the spinal cord following intravenous delivery of bone marrow cells. Glia 2002, 39, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, C.P.; Schwarz, E.J.; Hess, D.; Widenfalk, J.; El Manira, A.; Prockop, D.J.; Olson, L. Marrow stromal cells form guiding strands in the injured spinal cord and promote recovery. Proc. Natl. Acad. Sci. USA 2002, 99, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Zappia, E.; Casazza, S.; Pedemonte, E.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Frassoni, F.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Chen, J.; Cui, Y.; Lu, M.; Elias, S.B.; Mitchell, J.B.; Hammill, L.; Vanguri, P.; Chopp, M. Human bone marrow stromal cell treatment improves neurological functional recovery in EAE mice. Exp. Neurol. 2005, 195, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gerdoni, E.; Gallo, B.; Casazza, S.; Musio, S.; Bonanni, I.; Pedemonte, E.; Mantegazza, R.; Frassoni, F.; Mancardi, G.; Pedotti, R.; et al. Mesenchymal stem cells effectively modulate pathogenic immune response in experimental autoimmune encephalomyelitis. Ann. Neurol. 2007, 61, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kassis, I.; Grigoriadis, N.; Gowda-Kurkalli, B.; Mizrachi-Kol, R.; Ben-Hur, T.; Slavin, S.; Abramsky, O.; Karussis, D. Neuroprotection and immunomodulation with mesenchymal stem cells in chronic experimental autoimmune encephalomyelitis. Arch. Neurol. 2008, 65, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Chen, C.P.; Lin, S.Z.; Lee, Y.J.; Li, H. Efficient tracking of non-iron-labeled mesenchymal stem cells with serial mri in chronic stroke rats. Stroke 2007, 38, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Detante, O.; Valable, S.; de Fraipont, F.; Grillon, E.; Barbier, E.L.; Moisan, A.; Arnaud, J.; Moriscot, C.; Segebarth, C.; Hommel, M.; et al. Magnetic resonance imaging and fluorescence labeling of clinical-grade mesenchymal stem cells without impacting their phenotype: Study in a rat model of stroke. Stem Cells Transl. Med. 2012, 1, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Voulgari-Kokota, A.; Fairless, R.; Karamita, M.; Kyrargyri, V.; Tseveleki, V.; Evangelidou, M.; Delorme, B.; Charbord, P.; Diem, R.; Probert, L. Mesenchymal stem cells protect CNS neurons against glutamate excitotoxicity by inhibiting glutamate receptor expression and function. Exp. Neurol. 2012, 236, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Drago, D.; Cossetti, C.; Iraci, N.; Gaude, E.; Musco, G.; Bachi, A.; Pluchino, S. The stem cell secretome and its role in brain repair. Biochimie 2013, 95, 2271–2285. [Google Scholar] [CrossRef] [PubMed]
- Jadasz, J.J.; Kremer, D.; Gottle, P.; Tzekova, N.; Domke, J.; Rivera, F.J.; Adjaye, J.; Hartung, H.P.; Aigner, L.; Kury, P. Mesenchymal stem cell conditioning promotes rat oligodendroglial cell maturation. PLoS ONE 2013, 8, e71814. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Siebzehnrubl, F.A.; Kandasamy, M.; Couillard-Despres, S.; Caioni, M.; Poehler, A.M.; Berninger, B.; Sandner, B.; Bogdahn, U.; Goetz, M.; et al. Mesenchymal stem cells promote oligodendroglial differentiation in hippocampal slice cultures. Cell. Physiol. Biochem. 2009, 24, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Lennon, D.P.; Eaton, V.; Maier, K.; Caplan, A.I.; Miller, S.D.; Miller, R.H. Human bone marrow-derived mesenchymal stem cells induce Th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia 2009, 57, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.R.; Stoutenger, B.R.; Robinson, A.P.; Spees, J.L.; Prockop, D.J. Human stem/progenitor cells from bone marrow promote neurogenesis of endogenous neural stem cells in the hippocampus of mice. Proc. Natl. Acad. Sci. USA 2005, 102, 18171–18176. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Kemp, K.; Ginty, M.; Hares, K.; Mallam, E.; Scolding, N. Human bone marrow-derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Res. 2009, 3, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Isele, N.B.; Lee, H.S.; Landshamer, S.; Straube, A.; Padovan, C.S.; Plesnila, N.; Culmsee, C. Bone marrow stromal cells mediate protection through stimulation of PI3-K/AKT and MAPK signaling in neurons. Neuroch. Int. 2007, 50, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Lanza, C.; Morando, S.; Voci, A.; Canesi, L.; Principato, M.C.; Serpero, L.D.; Mancardi, G.; Uccelli, A.; Vergani, L. Neuroprotective mesenchymal stem cells are endowed with a potent antioxidant effect in vivo. J. Neurochem. 2009, 110, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Menge, T.; Zhao, Y.; Zhao, J.; Wataha, K.; Gerber, M.; Zhang, J.; Letourneau, P.; Redell, J.; Shen, L.; Wang, J.; et al. Mesenchymal stem cells regulate blood-brain barrier integrity through TIMP3 release after traumatic brain injury. Sci. Transl. Med. 2012, 4, 161ra150. [Google Scholar] [CrossRef] [PubMed]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Mohyeddin Bonab, M.; Yazdanbakhsh, S.; Lotfi, J.; Alimoghaddom, K.; Talebian, F.; Hooshmand, F.; Ghavamzadeh, A.; Nikbin, B. Does mesenchymal stem cell therapy help multiple sclerosis patients? Report of a pilot study. Iran. J. Immunol. 2007, 4, 50–57. [Google Scholar] [PubMed]
- Yamout, B.; Hourani, R.; Salti, H.; Barada, W.; El-Hajj, T.; Al-Kutoubi, A.; Herlopian, A.; Baz, E.K.; Mahfouz, R.; Khalil-Hamdan, R.; et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: A pilot study. J. neuroimmunol. 2010, 227, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Bang, O.Y.; Lee, J.S.; Lee, P.H.; Lee, G. Autologous mesenchymal stem cell transplantation in stroke patients. Ann. Neurol. 2005, 57, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Hong, J.M.; Moon, G.J.; Lee, P.H.; Ahn, Y.H.; Bang, O.Y. A long-term follow-up study of intravenous autologous mesenchymal stem cell transplantation in patients with ischemic stroke. Stem Cells 2010, 28, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Ra, J.C.; Shin, I.S.; Kim, S.H.; Kang, S.K.; Kang, B.C.; Lee, H.Y.; Kim, Y.J.; Jo, J.Y.; Yoon, E.J.; Choi, H.J.; et al. Safety of intravenous infusion of human adipose tissue-derived mesenchymal stem cells in animals and humans. Stem Cells Dev. 2011, 20, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.S.; Bar-Or, A.; Atkins, H.L.; Karussis, D.; Frassoni, F.; Lazarus, H.; Scolding, N.; Slavin, S.; Le Blanc, K.; Uccelli, A. The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: Consensus report of the international MSCT study group. Mult. Scler. 2010, 16, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Connick, P.; Kolappan, M.; Crawley, C.; Webber, D.J.; Patani, R.; Michell, A.W.; Du, M.Q.; Luan, S.L.; Altmann, D.R.; Thompson, A.J.; et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012, 11, 150–156. [Google Scholar] [CrossRef]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.W.; Behrens, M.I.; Chung, C.; Bhattacharyya, T.; Choi, D.W. Susceptibility to apoptosis is enhanced in immature cortical neurons. Brain Res. 1997, 759, 228–232. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.M.; Dahl, S.L. Recombinant human tumor necrosis factor receptor (p75) Fc fusion protein (TNFR:Fc) in rheumatoid arthritis. Ann. Pharmacother. 1997, 31, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Kucharova, K.; Chang, Y.; Boor, A.; Yong, V.W.; Stallcup, W.B. Reduced inflammation accompanies diminished myelin damage and repair in the NG2 null mouse spinal cord. J. Neuroinflamm. 2011, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.; Pitt, D.; Raine, C.S. Multiple sclerosis: Altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann. Neurol. 2001, 50, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Groom, A.; Zhu, B.; Turski, L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat. Med. 2000, 6, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Lennon, D.P.; Caplan, A.I.; DeChant, A.; Hecker, J.; Kranso, J.; Zaremba, A.; Miller, R.H. Hepatocyte growth factor mediates mesenchymal stem cell-induced recovery in multiple sclerosis models. Nat. Neurosci. 2012, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Reuss, B.; von Bohlen und Halbach, O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003, 313, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Valable, S.; Muller, G.J.; Roberts, M.L.; Divoux, D.; Tinel, A.; Voulgari-Kokota, A.; Tseveleki, V.; Altruda, F.; Lassmann, H.; et al. FLIP(L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J. Neurosci. 2007, 27, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, S.; Quattrini, A.; Brambilla, E.; Gritti, A.; Salani, G.; Dina, G.; Galli, R.; Del Carro, U.; Amadio, S.; Bergami, A.; et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 2003, 422, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Butti, E.; Bacigaluppi, M.; Rossi, S.; Cambiaghi, M.; Bari, M.; Cebrian Silla, A.; Brambilla, E.; Musella, A.; De Ceglia, R.; Teneud, L.; et al. Subventricular zone neural progenitors protect striatal neurons from glutamatergic excitotoxicity. Brain 2012, 135, 3320–3335. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Hawthorne, A.L.; Bai, L.; Miller, R.H.; Silver, J. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J. Neurosci. 2010, 30, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Meirelles, L.; de Deus Wagatsuma, V.M.; Malta, T.M.; Bonini Palma, P.V.; Araujo, A.G.; Panepucci, R.A.; Silva, W.A.; Kashima, S.; Covas, D.T. The gene expression profile of non-cultured, highly purified human adipose tissue pericytes: Transcriptomic evidence that pericytes are stem cells in human adipose tissue. Exp. Cell Res. 2016, 349, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.G. The role of excitotoxic programmed necrosis in acute brain injury. Comput. Struct. Biotechnol. J. 2015, 13, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Matthews, N.; Neale, M.L. Cytotoxicity assays for tumour necrosis factor and lymphotoxin. In Lymphokines & Interferons, a Practical Approach; Clemens, M.J., Morris, A.G., Gearing, A.J.H., Eds.; IRL Press: Oxford, UK, 1987; pp. 221–225. [Google Scholar]
- Lipp, P.; Niggli, E. Ratiometric confocal Ca(2+)-measurements with visible wavelength indicators in isolated cardiac myocytes. Cell Calcium 1993, 14, 359–372. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papazian, I.; Kyrargyri, V.; Evangelidou, M.; Voulgari-Kokota, A.; Probert, L. Mesenchymal Stem Cell Protection of Neurons against Glutamate Excitotoxicity Involves Reduction of NMDA-Triggered Calcium Responses and Surface GluR1, and Is Partly Mediated by TNF. Int. J. Mol. Sci. 2018, 19, 651. https://doi.org/10.3390/ijms19030651
Papazian I, Kyrargyri V, Evangelidou M, Voulgari-Kokota A, Probert L. Mesenchymal Stem Cell Protection of Neurons against Glutamate Excitotoxicity Involves Reduction of NMDA-Triggered Calcium Responses and Surface GluR1, and Is Partly Mediated by TNF. International Journal of Molecular Sciences. 2018; 19(3):651. https://doi.org/10.3390/ijms19030651
Chicago/Turabian StylePapazian, Irini, Vasiliki Kyrargyri, Maria Evangelidou, Anda Voulgari-Kokota, and Lesley Probert. 2018. "Mesenchymal Stem Cell Protection of Neurons against Glutamate Excitotoxicity Involves Reduction of NMDA-Triggered Calcium Responses and Surface GluR1, and Is Partly Mediated by TNF" International Journal of Molecular Sciences 19, no. 3: 651. https://doi.org/10.3390/ijms19030651
APA StylePapazian, I., Kyrargyri, V., Evangelidou, M., Voulgari-Kokota, A., & Probert, L. (2018). Mesenchymal Stem Cell Protection of Neurons against Glutamate Excitotoxicity Involves Reduction of NMDA-Triggered Calcium Responses and Surface GluR1, and Is Partly Mediated by TNF. International Journal of Molecular Sciences, 19(3), 651. https://doi.org/10.3390/ijms19030651