1. Introduction
Estrogen Receptor α (ERα) is the key transcriptional factor of the luminal breast cancer subtype, which comprises three quarter of breast cancer cases [
1], by mediating estrogen signaling in breast cancer cells. Nowadays, current endocrine treatments that block estrogen action include Selective Estrogen-Receptor Modulators (SERMs), Fulvestrant (pure anti-estrogen that degrades ERα protein) and Aromatase Inhibitors (AI) [
2]. SERMs are synthetic compounds that function as agonists or antagonists for estrogen receptors in a tissue-specific manner. The first SERM that has been used successfully in the clinic is tamoxifen (TAM) which functions as cell type-specific anti-estrogens [
3]. The most common endocrine treatments today are AIs, which block the biosynthesis of estrogens [
4]. However, this therapy fails in one-fourth of ERα+ breast cancer cases because of de-novo or acquired endocrine resistance, which is generally characterized by accelerated tumor growth and increased aggressive behavior [
5].
ERα regulates biological processes in breast cancer cells by ligand-dependent (estrogen-ERα complex) or ligand-independent action (apoERα) [
6]. Whereas estrogen dependent signaling was deeply studied in the last decades [
7], few works were based on understanding apoERα mechanism in BC. It was reported that apoERα acts in cooperation with FOXA1 and AP2γ to maintain the luminal epithelial enhancer landscape and when one of these factors are suppressed, enhancers progressively collapse [
6]. As previously demonstrated for E2-bound ERα genomic action [
8], gene expression regulation by apoERα seems dependent on long range interaction between apoERα-bound enhancers and promoters.
Recently, Super-Enhancers (SEs) emerged as a novel class of distal
cis regulatory elements, defined as large clusters of putative enhancers in close genomic proximity which drive gene expression to define cell lineage identity [
9]. SEs tend to span large genomic regions with unusually strong enrichment of lineage-specific TFs and co-activators binding. These stitched enhancers have additive and synergistic functions allowing them to drive high levels of tissue-specific gene expression. Commonly, SEs were predicted by identification of genomic regions enriched in H3K27ac ChIP-Seq signal compared to input or control experiment [
10]. SEs represent the core of the transcription regulation for cell-type-specific gene expression and play a key role in cellular development and dynamic response to environmental stimuli [
11,
12]. Deciphering the active SEs in a specific context is helpful for the identification of key
cis-regulation of coding and non-coding genes expression [
13]. In MCF-7 cells, it was recently demonstrated that ERα is required for SEs formation upon binding to canonical response elements and in presence of FoxA1 pioneering factor [
14]. This study elucidated SEs assembly in an estrogen-dependent manner, but the mechanism of SEs modulation by ligand-independent ERα remains unclear.
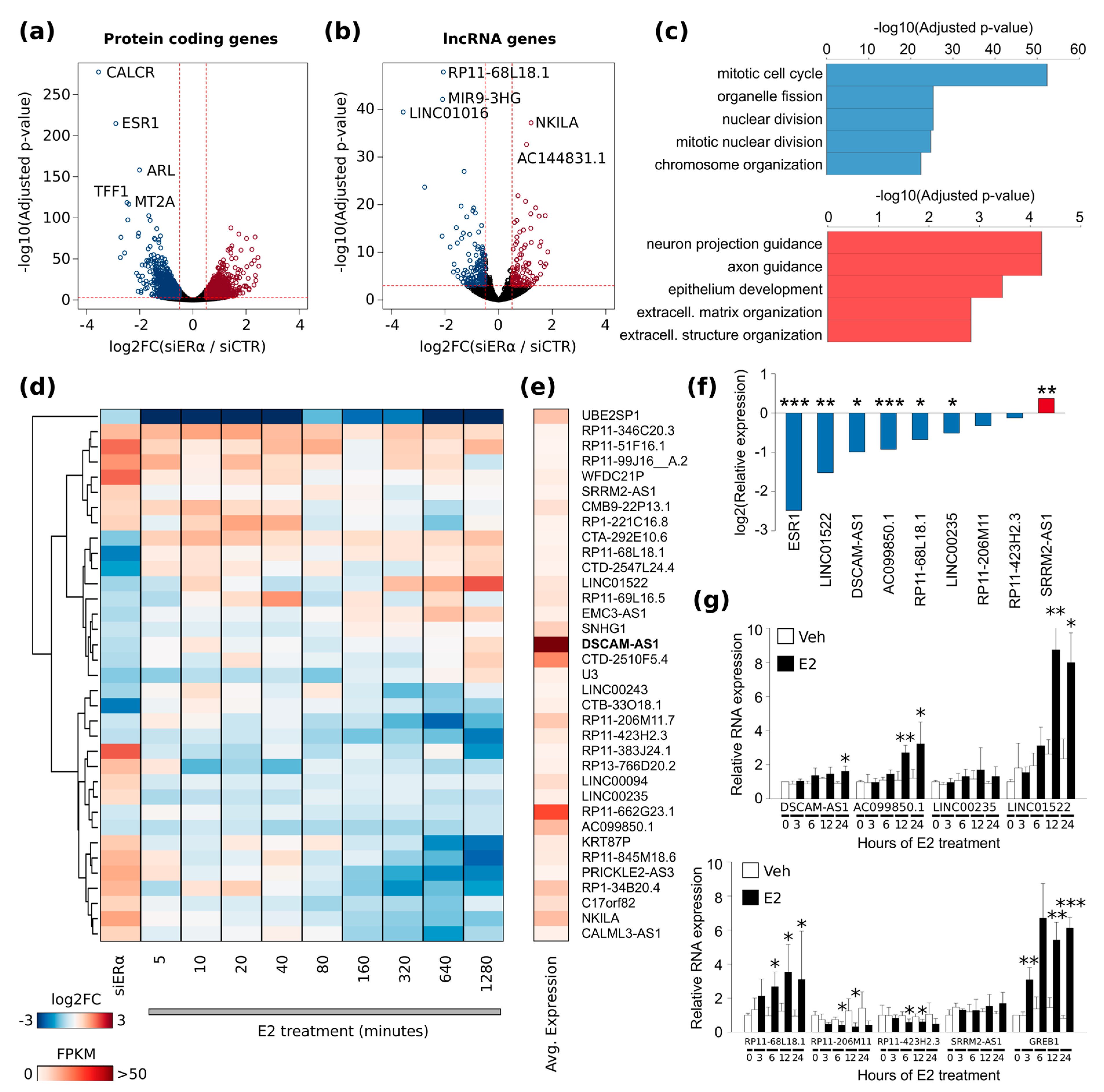
A full collection of lncRNAs has emerged during the last decade, stressing on their importance as regulators of cellular development and differentiation. They are expressed at lower levels than protein-coding transcripts, but compared to the latter, their pattern of expression is remarkably cell type-specific [
15,
16]. Indeed, to achieve a comprehensive annotation, lncRNAs expression has been quantitatively analyzed in several tissues and cell types by high-throughput RNA sequencing (RNA-seq) [
17]. In BC research, lncRNAs have been involved in regulation of mammary epithelial cells homeostasis [
18] and several studies focused on lncRNAs regulated by estrogen [
8,
19,
20]. Finally, we recently identified for the first time a set of apoERα-regulated lncRNAs [
21]. Comparison with publicly available data showed that these lncRNAs are highly specific for the luminal BC subtype and therefore very promising in defining peculiar BC features. Among this set,
DSCAM-AS1 is the most abundantly expressed lncRNA in BC cell lines and tissue samples. Expression of
DSCAM-AS1 has a very peculiar dependence over ERα: the increased ERα binding at
DSCAM-AS1 promoter upon estradiol stimulus does not lead to a concordant up-regulation of gene transcription over at least 24 h indicating a necessary and sufficient apoERα action for the regulation of
DSCAM-AS1 transcription [
21]. It has been reported that ERα binding to
DSCAM-AS1 promoter is high in tumor tissues from patients unresponsive to Tamoxifen treatment and that
DSCAM-AS1 expression is upregulated in Tamoxifen-resistant cellular models [
22], contrary to typical estrogen-responsive genes that are strongly downregulated. These results suggest that actually distal putative enhancer or SE regions might be responsible for this peculiar transcriptional regulation and might be more generally involved in transcriptional regulation of cell-specific lncRNAs.
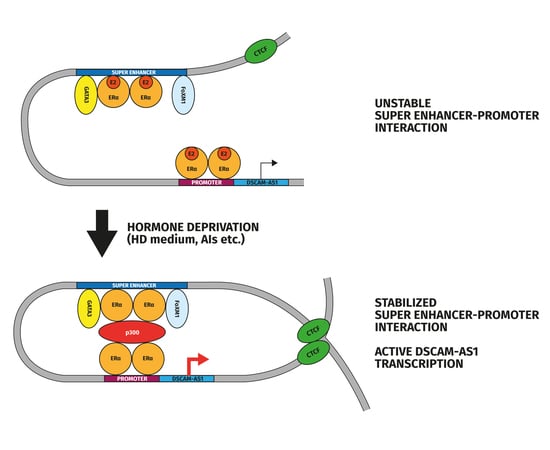
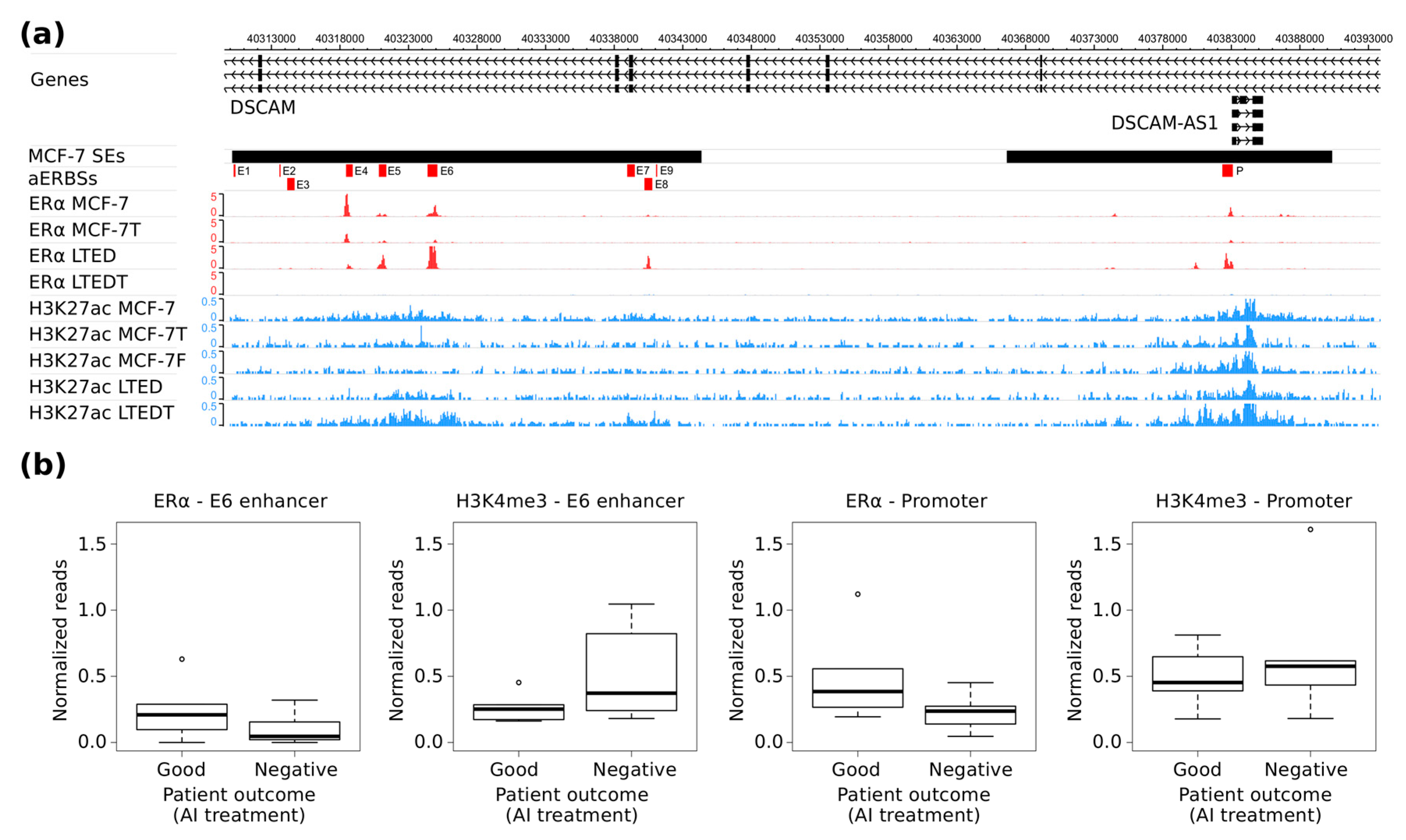
In the present study, we extended the analysis of apoERα-regulated transcriptome in MCF-7, characterized apoERα binding at SEs (SE-aERBSs) and defined apoERα-regulated lncRNAs that are associated and regulated by SE activation. In particular, we focused our attention on DSCAM-AS1 associated SE which is stabilized in a hormone-independent manner and whose regulation depends on the presence of apoERα. It represents a paradigmatic example of how luminal lncRNAs are regulated by specific SEs activated through unliganded ERα, which is extremely important to understand luminal breast tumor growth and progression, in view of the fact that common endocrine treatments today (AIs) deplete the organism of estrogenic hormones.
3. Discussion
In this work, we have presented novel results that help understanding the role of Estrogen Receptor α in absence of hormones (apoERα), which is a critical issue for Breast Cancer (BC) patients given adjuvant treatment with Aromatase Inhibitors (AIs). We performed a deep triplicate RNA-Seq in hormone-depleted MCF-7 cells upon ERα silencing and, specifically, we were interested to understand whether Super Enhancers (SEs) were involved in sustaining the basal expression of a group of luminal-specific lncRNAs. Our study indicates that a set of apoERα dependent lncRNAs lie close to SEs, suggesting a strict dependence on these regions. Focusing on a representative lncRNA gene, DSCAM-AS1, we observed that aERBSs within the major SE in this region are active enhancers, also displaying long-distance contacts with the DSCAM-AS1 promoter, even in the absence of hormones. Moreover, our data suggests that the chromatin domain confining this interaction may depend upon the estrogen-independent CTCF binding in proximity of DSCAM-AS1. Finally, comparison of SE activity in different BC cell lines revealed a relationship between ERα expression level and SE activity as well as DSCAM-AS1 responsiveness to E2 treatment. Noteworthy, we confirmed the active epigenetic status of DSCAM-AS1 locus in BC patients unresponsive to AI, but low ERα binding at SE was observed in these samples.
The identification of SEs helped us to understand the peculiar transcriptional program regulating cell-type specific gene expression. Recently, several evidences demonstrated that cell-specific enhancers play an important role in tumorigenesis [
11]. Indeed, the study of SEs driving the expression of specific cancer associated lncRNAs may delineate an additional informative layer on cancer development.
One important limitation of our results is represented by the fact that the possible targets of SE were identified only based on the distance, i.e., we attributed to each SE the closest apoERα-regulated lncRNA gene. While on the basis of the reported frequency of enhancer-promoter interaction this is justified, the ENCODE project and other studies have reported that in a number of cases enhancers interact with promoters jumping over several genes [
26]. Clearly, the matter would require direct long-range interaction studies using the Hi-C technology [
33] and derivatives. Nevertheless, at least in the case of
DSCAM-AS1, the prediction appeared correct and could be confirmed by our ChIA-PET network model.
The expression of the carcinoma-specific
DSCAM-AS1 is particularly interesting due to its abundance and close association with unliganded ERα activity.
DSCAM-AS1 loss impairs indeed BC cells proliferation and its expression is somehow implicated in Tamoxifen-resistance [
21,
22]. Here, we confirmed and extended previous results of our group [
21] by validating ERα binding both in the promoter region and in putative enhancer elements that was very high in absence of estrogen, demonstrating only a slight increase after estradiol stimulation, and confirmed that this increase is not paralleled by transcriptional activation, whereas down-regulation of ERα by RNA interference led to significant transcriptional inhibition. On the contrary, low levels of the coding DSCAM transcript are detectable in breast cancer cells and no function of DSCAM in mammary tissues has been described to date. DSCAM mRNA slightly decreases upon ERα-silencing but it appears induced by estrogen [
21,
34], suggesting that the estrogen-induced re-localization of aERBSs might differentially regulate the expression of these sense and antisense transcripts in BC cells. Altogether, this data indicates a specific ligand-independent ERα program to sustain
DSCAM-AS1 expression in BC cells.
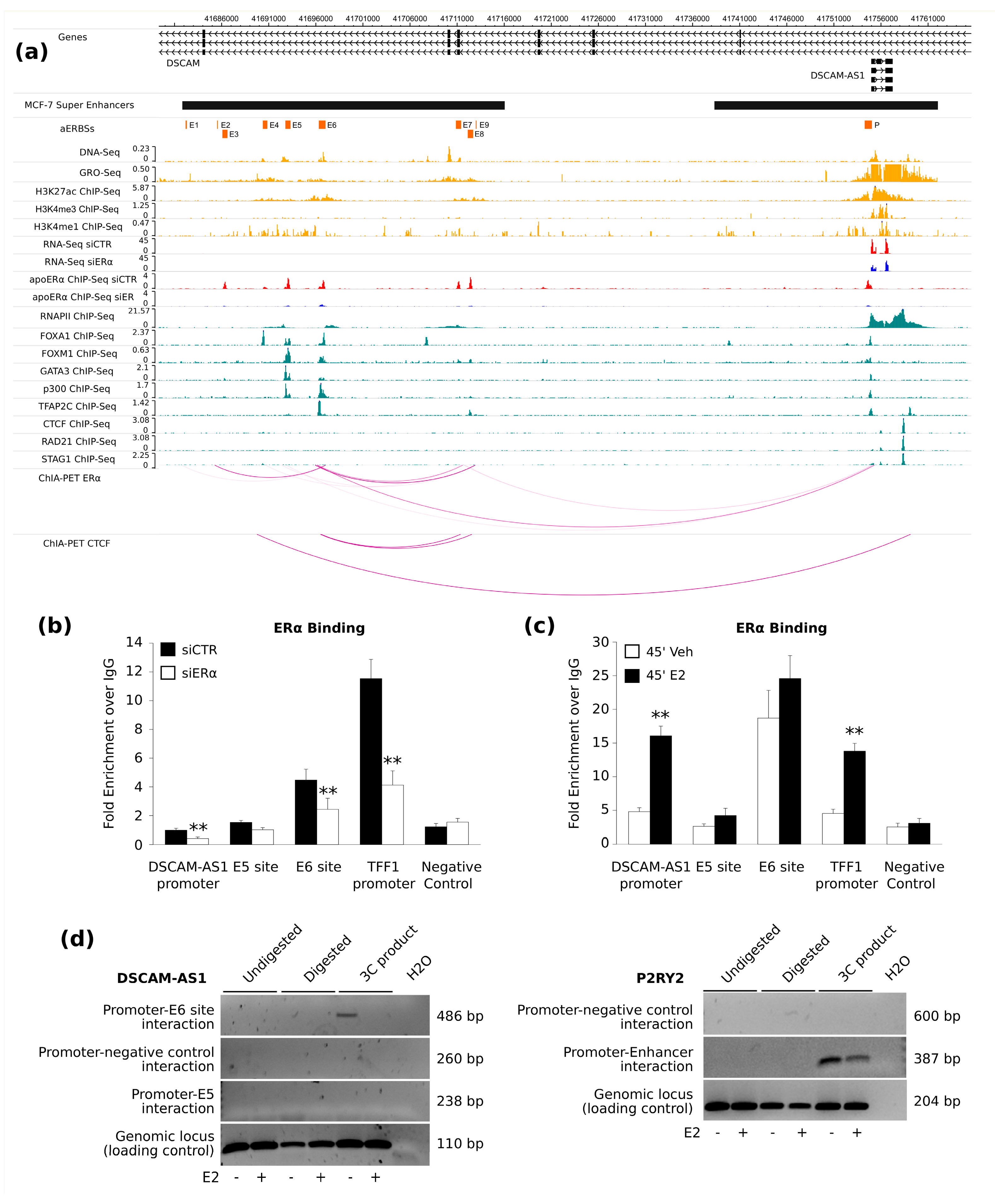
The presence of apoERα bound to an intronic element, which can increase quantitatively in response to estrogen but not transcriptionally, is not novel: a similar situation was reported for an intronic site in the retinoic acid receptor alpha 1 gene (
RARA) [
35] suggesting that such regulatory modality may be more frequent in lncRNA genes, but also present in protein-coding genes. Notably, also the
RARA gene has a very important developmental function in epithelial mammary cells. One important novel finding in our data is represented by the state of activation of the aERBS within the major SE upstream the
DSCAM-AS1 gene. We evaluated p300 enrichment confirming the active status of these regulatory regions. In particular, the transcriptional coactivator p300 possesses intrinsic acetyltransferase activity and it was shown to play a critical role in transcriptional regulation [
36]. Consistent with our results, p300 occupancy is a highly accurate method to identify enhancers and their associated activities [
37,
38]. Interestingly, we observed that ERα depletion induced a decrease of p300 occupancy, suggesting that the activity of these enhancers requires the presence of ERα. Moreover, p300 binding to the
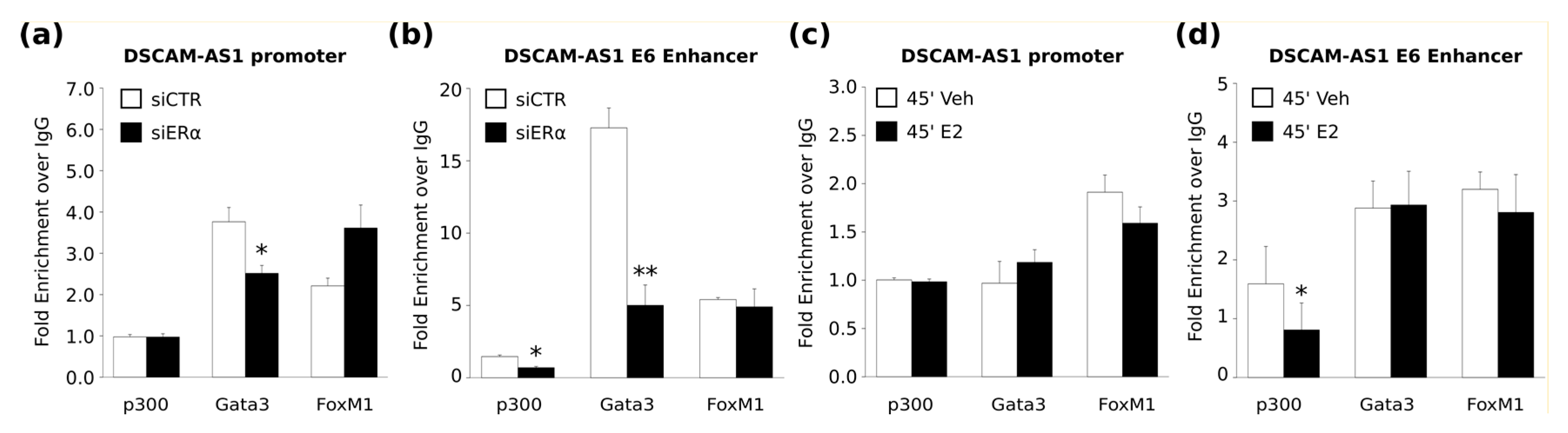
DSCAM-AS1 enhancer is conserved rather in hormone-deprived medium than upon estrogen treatment, indicating that this SE is not an early estrogen target site. However, the dynamic distribution of p300 in BC cells in the presence or absence of estrogenic hormones and its effect on the prognosis of BC are poorly understood [
38].
Our analysis confirms the model where apoERα collaborates with other TFs to maintain the luminal epithelial enhancer landscape [
6]. These cooperation are underlined by the cross-regulatory loop involving GATA-3 and ERα or AP2γ and ERα, which sustain each other’s expression in BC cells [
39,
40]. Interestingly, we also observed that ERα depletion induced a decrease of GATA3 occupancy; therefore, the regulation of
DSCAM-AS1 gene expression seems to be based on long range interaction between apoERα-bound enhancer and the gene promoter as previously described for some estrogen-dependent ERα target genes such as
TFF1 and
GREB1 [
41]. Noteworthy, our analysis suggests that only the E6 aERBS is involved in
DSCAM-AS1 regulation. In fact, both ERα binding and 3C analysis show very low activity of E5, also reflecting public ChIA-PET data of MCF-7 cells grown in full medium, i.e., under low hormone concentrations [
26].
Therefore, we attempted DSCAM-AS1 promoter-enhancer interaction before and after estrogen stimulation and we could detect at least the E6-promoter interaction, further demonstrating that this enhancer is active even in the absence of estrogen. However, it should be emphasized that this is a preliminary and qualitative result, which should be confirmed by further experiments to quantify the frequency of DSCAM-AS1 promoter-enhancer interaction in different experimental conditions. This data will allow understanding if this interaction is more favored or stabilized in absence of hormones, as we hypothesize.
The eukaryotic genome is organized into functionally and structurally distinct chromatin domains which orchestrate spatial and temporal gene regulation. CTCF, the most characterized insulator-binding protein, is directly involved in chromatin architecture by the formation of barriers which limit the diffusion of chromatin states and promote tissue-specific local chromatin hubs. CTCF binding to insulator sequences can partition the genome into distinct ERα-regulatory blocks [
42] defining sensitive and insensitive regions [
43]. However, redistribution of CTCF binding sites after E2 treatment was not generally observed [
44]. Since CTCF binding sites were mapped around
DSCAM-AS1 and a long-range interaction was observed between a site close to
DSCAM-AS1 3’ end and a site farther upstream the SE, we asked whether CTCF binding could justify the peculiar mode of action of the
DSCAM-AS1 enhancer/promoter regulatory system. Noteworthy, we found that CTCF occupancy upstream
DSCAM-AS1 decreases after ERα silencing in HD medium, whereas it decreases significantly downstream the
DSCAM-AS1 gene region after estradiol stimulation, suggesting that estrogen, or its absence, may induce a redistribution of long-range interactions. Taken together, these results suggest a non-trivial regulatory model at this site, which may involve channeling SE action on the
DSCAM-AS1 promoter in cancer cells overexpressing ERα due to strict domain definition by CTCF. We also supposed that the reduction of CTCF signal upon estradiol stimulation indicate that these enhancers might regulate distal genes surmounting the protein insulator. Interestingly, the analysis of public RNA-Seq data MCF-7 transfected with siCTCF and treated with E2 for 3 h show a reduction of
DSCAM-AS1 level in siCTCF-treated cells (
Supplementary Figure S2e).
Our data suggest a model in which CTCF protein acts by insulating the interaction between
DSCAM-AS1 and associated SE. Within the SE a hierarchical ERα binding starting from E6 aERBS maintains the long-range chromatin contact even in hormone-deprived cells. A similar hierarchical model was recently proposed by different groups [
14,
45], however, if this model can apply to the regulation of other ERα-dependent lncRNAs is not known at present and will require extension of these studies in the near future.
Finally, analysis of the
DSCAM-AS1 locus in several BC cell lines suggested a direct correlation between the level of ERα expression and the
DSCAM-AS1 SE activation status. Indeed, in MCF-7, (the BC cell line characterized by the highest ERα expression), the
DSCAM-AS1 SE is completely active, while only the
DSCAM-AS1 promoter region is active in ZR-75.1 (characterized by a lower ERα expression). Furthermore, the LTED cell model system, which is characterized by an elevated ERα expression [
46], were also characterized by the highest ERα SE occupancy among all the cell lines analyzed. However, given the extensive genetic and epigenetic heterogeneity driving the acquisition of a hormone-independent phenotype in the different LTED models [
31], limited general conclusion can be translated to the clinical settings.
Analysis of ERα and H3K4me3 ChIP-Seq data clearly show extensive heterogeneity among patients. Using these data, we were not able to confirm a strong ERα binding at
DSCAM-AS1 SE, but we clearly observed that
DSCAM-AS1 locus is active in four out six patients non-responsive to AI treatments. Since patients with a good prognosis present no H3K4me3 signal at DSCAM-AS-SE, its activated-status is correlated with a negative prognosis, suggesting it as epigenetic-predictor of AI-treatment response. As far as the other common endocrine treatment is concerned, i.e., Tamoxifen, Niknafs and coworkers reported that Tamoxifen-resistant patients display ERα binding to
DSCAM-AS1 promoter, as opposed to Tamoxifen responders [
22]. Using the same data [
47], we additionally examined ERα binding at the
DSCAM-AS1 SE, confirming that ERα binding is limited to non-responder patients (
Supplementary Figure S2d,f). Altogether, these data support the fact that
DSCAM-AS1 does not behave as a typical estrogen-responsive gene, since its active transcriptional status is maintained both in absence of estrogen (AI) and in the presence of antagonistic ERα ligands (Tamoxifen).
Moreover,
DSCAM-AS1 genes was included in the gene classifier of drug responsiveness proposed by Jansen and colleagues [
32].
In our lab, we are actively exploring DSCAM-AS1 expression in tissues of BC patients to demonstrate it possible role as hormone-responsiveness marker. At the same time, several other lncRNA loci are being studied to understand unliganded ERα action in luminal breast cancer.
4. Materials and Methods
4.1. Cells Culture and Treatment
MCF-7 and ZR-75.1 cells were routinely grown in DMEM medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 50 U/mL penicillin and 50 μg/mL streptomycin. T-47D cells were routinely grown in RPMI medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 50 U/mL penicillin and 50 μg/mL streptomycin. Hormone-deprived medium (HD) was obtained from phenol red-free DMEM supplemented with 5% charcoal-dextran-treated serum. MCF-7 cells were grown at least 4 days in HD medium before 17β-estradiol treatment (E2). 17β-estradiol (Sigma -Aldrich, St. Louis, MO, USA) was added at a final concentration of 10–8 M.
4.2. Small Interfering RNA (siRNA)
MCF-7 cells were grown in HD medium (for two days in this conditioned medium) before being transfected with siRNAs using Lipofectamine2000 (Life Technologies, Carlsbad, CA, USA), according to the reverse transfection protocol. Stealth RNAi™ siRNAs from Invitrogen were used to target ERα mRNA (ESR1HSS103376, ESR1HSS103377, ESR1HSS176619); stealth RNAi™ siRNA Negative Control medium GC was used as a control (siCTR). Any experiment was carried out 48 h after siRNA transfection in order to achieve the maximum efficiency of RNA interference.
4.3. RNA-Seq
MCF-7 cells were grown for 2 days in HD medium before siRNAs transfection (siCTR or siERa). RNA was harvested after 48 h from transfection by RNA isolation kit (RNeasy Mini Kit, Qiagen 74104). RNA quality check (RNA integrity number (RIN) > 8) was achieved with Fragment Analyzer (Advanced Analytical Technologies, Inc, Ankeny, IA, USA) and quantified with Qubit (Qubit™ RNA HS Assay Kit, ThermoFisher Scientific, Waltham, MA, USA;Q32852). Two μg of RNA were polyA+ selected and RNA-Seq libraries were constructed using Illumina TruSeq RNA sample prep kit (TruSeq™ RNA Sample Prep Kit v2-Set B, Illumina, RS-122-2002). Paired-end (PE) cluster generation was performed using cBot on Flow Cell v3 (TruSeq PE Cluster Kit v3-cBot-HS, Illumina, PE-401-3001). Sequencing of libraries was performed on the HiSeq sequencing system (Illumina, San Diego, CA, USA).
Raw and processed RNA-Seq data were deposited at GSE108693.
4.4. RNA-Seq Data Analysis
Raw RNA-Seq reads were aligned using
TopHat v2 [
48]. Gencode v24 was used as reference set of gene annotations. Read count was performed using
featureCounts algorithm [
49] and read count tables were normalized with
DESeq2 package [
50]. Normalized read counts were converted to fragments per kilobase of exons per million fragments mapped (FPKM) considering the length of the longest isoform of each gene and the millions of reads counted by featureCounts.
Differentially expression between siCTR and siERα-treated MCF-7 was performed using
DESeq2 in default settings. A gene was considered as differentially expressed if associated with an adjusted
p-value < 0.001. To identify genes differentially expressed in the time-course E2-treatment experiment,
maSigPro R package [
51] was applied in default setting by considering the adjusted
p-value computed using the p.vector function of the package. A gene was considered as differentially expressed if associated with an adjusted
p-value < 0.05. Gene to SEs association was performed by computing the distance between gene TSSs and each SE center. Only genes mapped within 100 kbp from a SE region were considered for the analysis.
Gene set functional analysis was performed using Enrichr algorithm [
52].
Analysis of GRO-Seq datasets from GSE45822 was performed using Bowtie v2.1.0 [
53] in default settings and the
local option.
4.5. Super Enhancer Prediction
A H3K27ac ChIP-Seq dataset performed on 48-h hormone-deprived MCF-7 cells treated with vehicle (GSM986079) was used for the identification of SE regions. Raw ChIP-Seq reads were aligned using Bowtie 2.0 algorithm in default settings [
53]. Significant H3K27ac ChIP-Seq peaks were called with MACS 2.1.0 [
54]. The input ChIP-Seq experiment was used as background for the analysis (GSM986091). These peaks were used as input for the ROSE algorithm [
24] analysis applied in default settings. Only SEs overlapped against apoERα ChIP-Seq peaks defined in [
6] were selected for the analysis. The set of SEs predicted in other BC cell lines were retrieved from [
30] supplemental material. These SE coordinates were converted in hg38 using LiftOver algorithm (
https://genome.ucsc.edu/cgi-bin/hgLiftOver).
4.6. Long Range Chromatin Interaction Network
The network model of Gene-aERBSs chromatin interaction was generated using the ChIA-PET data from GSE39495. Each pair of interacting genomic regions was overlapped with the aERBSs and gene promoter coordinates. A promoter was defined as a region of −2 kbp and +500 bp around the gene TSS. Each aERBS was equally extended by 1 kbp in each size. The average number of interacting region between each aERBS-gene, gene-gene, or aERBS-aERBS pair was computed and represented as link width between the connected nodes. Gene were color-coded according to their expression log2FC in the siERα RNA-Seq experiment while aERBS nodes were colored according to their epigenetic classification defined in [
27].
4.7. DSCAM-AS1 Locus Analysis
The analysis of
DSCAM-AS1 locus and upstream enhancer regions was performed using the Washu Genome Browser [
55]. ChIP-Seq genomic signals from public experiments performed in hormone-deprived MCF-7 were defined by realignment of raw ChIP-Seq reads using Bowtie 2.0 algorithm in default settings. Then, alignment bam files were converted to bedgraph files using BEDTools algorithm [
56]. Each ChIP-Seq genomic signal was converted in count per millions by dividing the read coverage data by the million number of sequencing reads in each experiment. In the analysis data from a ChIP-Seq performed against H3K27ac (GSM1115993), H3K4me3 (GSM1382470), H3K4me1 (GSM1382469), RNAPII (GSM1116656), FoxA1 (GSM588929), FoxM1 (GSM1001003), Gata3 (GSM986074), p300 (ERR045733), AP2γ (GSM588927), CTCF (GSM822308), Rad21 (GSM614613), and Stag1 (GSM614616) were considered. ERα ChIP-Seq and RNA-Seq data from siCTR or siERα-transfected MCF-7 (GSE53533) were also considered in the analysis. DNase-seq were obtained from GSM1024764. ERα and H3K4me3 ChIP-Seq data from AI-treated patients were obtained from GSE40867 while data from Tamoxifen-treated patients were retrieved from GSE32222.
Data from ENCODE long-range chromatin interaction experiments were analyzed by considering the ERα and CTCF ChIA-PET experiments performed in complete-medium grown MCF-7. The processed data of these experiments were provided by the public track hub “long-range chromatin interaction experiments” of the Washu Genome Browser.
4.8. Protein EXTract and Western Blot Analysis
Whole-cell lysate was harvested from organic-phenol phase after RNA purification in Purezol™ reagent (Bio-Rad, Hercules, CA, USA). 70 μg of protein extracts were denatured by boiling in 6x Loading Dye, fractionated by SDS-PAGE electrophoresis in 6% polyacrylamide gel and finally transferred on PVDF membrane (Millipore). Antibodies used were designed against ERα (Santa Cruz Biotechnology, Dallas, TX, USA; sc543 and sc-7207), CTCF (Merck-Millipore, Burlington, MA, USA; 07-729), GATA3 (Santa Cruz Biotechnology, Dallas, TX, USA; sc-268), p300 (Santa Cruz Biotechnology Dallas, TX, USA sc-585), FOXM1 (Santa Cruz Biotechnology Dallas, TX, USA sc-376471) and Hsp90 (Cell Signaling Technology, Danvers, MA, USA; 4874) proteins. Proteins quantitation was performed with Volume Analysis Tool from Quantity One™ software, version 4.6.6 (Bio-Rad).
4.9. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
RNA was isolated from MCF-7 cells using Purezol™ reagent (Bio-Rad). All total isolated RNAs were subjected to DNase treatment to remove contaminating genomic DNA (ezDNase™ Enzyme, Invitrogen, Carlsbad, CA, USA; 11766051). First strand cDNA synthesis from 250 ng of total RNA template was performed with SensiFAST cDNA Synthesis Kit (Bioline, London, UK; BIO-65054), followed by SYBR-green qRT-PCR amplification (iTaq UniverSYBR Green, Bio-Rad 1725124). Real-time PCR primers for human 18S, ERα, GREB1 RNAs were purchased from Qiagen (Hilden, Germany; QuantiTect™ Primer Assay). Custom expression-primer pairs are reported in
Supplemental Table S6.
4.10. Chromatin Immunoprecipitation Assay (ChIP)
MCF-7 cells were grown for 2 days in HD medium before siRNAs transfection or 45 min of E2 treatment. ChIP experiments were then performed as follows: cells were cross-linked by addition of formaldehyde at 1% final concentration (Formaldehyde solution, Sigma-Aldrich 252549) and incubated 10 min at 37 °C. Cross-linking was stopped by addition of glycine solution to a final concentration of 0.125 M for 5 min on a shaker at room temperature. Cross-linked cells were then washed twice with ice-cold PBS supplemented with complete protease inhibitors cocktail and collect by scraping (Protease Inhibitor Cocktail, Sigma-Aldrich P2714-1). Cell pellets were subjected to lysis on ice for 10 min with Lysis Buffer 1 (5 mM Pipes pH 8, 85 mM KCl, 0.5 % NP40) supplemented with complete protease inhibitors cocktail. Subsequently, nuclei pellets, obtained by a 5 min spin cycle at 4 °C (4000 rpm) were exposed once again to 10 min lysis in Lysis Buffer 2 (1% SDS, 5 mM EDTA, 50 mM Tris-HCl pH 8.1) supplemented with complete protease inhibitors cocktail. Total extracted chromatin was sonicated to an average size of 250–500 bp by using an immersion sonicator device. The desired fragments size was checked on 1.2% agarose-gel and quantified by Nanodrop spectrophotometer at 280 nm, in order to use 50 μg of chromatin per IP. 10 μL (1%) of chromatin extracts was recovered as input normalization-control for each experimental condition. Chromatin extracts were diluted with IP-buffer (1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl of pH 8.1, 150 mM NaCl, supplemented with complete protease inhibitors) and incubated overnight with the specific antibody or IgG at 4° C on a rotating platform. Protein A and G sepharose-beads (GE Healthcare Life Sciences; Little Chalfont, UK; 17-5280-01 and 17-0618-01 respectively) were pre-coated with IP buffer supplemented with 5% BSA, in order to reduce nonspecific antibody binding. Upon 2 h of Protein A or G sepharose-beads incubation (depending on antibody source, i.e., rabbit or mouse respectively), samples were washed sequentially for 5 min, on a rotating platform with 1 mL of three different Washing Buffer (Washing buffer 1: 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris HCl pH 8, 150 mM NaCl; Washing buffer 2 : 0.1 SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris HCl pH 8, 500 mM NaCl; Washing buffer 3: 0.25 M LiCl, 1% NP40, 1% Na DOC, 1 mM EDTA, 10 mM Tris HCl pH8) and twice with TE buffer (10 mM Tris HCl pH8, 1 mM EDTA). After complexes elution at RT with elution buffer (1% SDS, 0.1 M NaHCO3), DNA fragments were de-crosslinked at 65 °C overnight with NaCl 5 M and by 1 h of proteinase K treatment. DNA purification was achieved with Phenol:Chloroform:IAA (25:24:1 UltraPure™ formulation, Ambion AM9730) according to the manufacturer’s instructions. Quantitative Real-time PCR was carried out on ChIP-enriched DNA using SYBR Green Master Mix. ChIP enrichment was normalized on input samples (1% of total chromatin used per IP) and expressed as fold-enrichment of specific binding over the control nonspecific IgG binding. Antibodies against human ERα (Santa Cruz Biotechnology, Dallas, TX, USA; sc534X, sc7207X), p300 (Santa Cruz Biotechnology, Dallas, TX, USA; sc-585X), CTCF (Merk-Millipore, Burlington, MA, USA; 07-729), GATA3 (Santa Cruz Biotechnology, Dallas, TX, USA; sc-268X), FOXM1 (Santa Cruz Biotechnology, Dallas, TX, USA; sc-376471X) and normal rabbit IgG (Merk-Millipore, Burlington, MA, USA; 12-370) were used in this assay. Custom ChIP primer pairs are reported in
Supplementary Table S6.
4.11. Qualitative Chromosome Conformation Capture (3C-PCR)
MCF-7 cells were grown for 2 days in HD medium before 45 min of E2 treatment. 3C procedure was performed essentially as described by [
8] with some modifications: Step: Crosslinking reaction. As well as for ChIP experiments, cells were cross-linked by addition of formaldehyde (1% final concentration) and incubated 10 min at 37 °C. Cross-linking was stopped by addition of glycine solution to a final concentration of 0.125 M for 5 min on a shaker at room temperature. Cross-linked cells were then washed twice with ice-cold PBS supplemented with complete protease inhibitors cocktail and collect by scraping. Cell pellets were subjected to lysis on ice for 10 min with ChIP Lysis Buffer 1 supplemented with complete protease inhibitors cocktail. Subsequently, nuclei pellets, obtained by a 5 min spin cycle at 4 °C (4000 rpm) were resuspended with 100 μL of 3C buffer (50 mM Tris-HCl pH 8.0; 50 mM NaCl; 10 mM MgCl2; 1 mM DTT). Step 2: Permeabilization and digestion. SDS (final concentration 0.2%) was added and samples were incubated at 37 °C for 1 h while shaking at 300 rpm. Then, Triton X-100, diluted in 1X ligase buffer (NEB, B0202S), was added at 1.2% final concentration and samples were incubated at 37 °C for 1 h while shaking at 300 rpm. A 10 μL aliquot was taken by pipetting on the wall of the tube (without mixing before). This sample corresponds to undigested DNA. DNA digestion will be done by adding a total of 450 U of restriction enzyme (BglII and NsiI, NEB R0144L and R0127L respectively) overnight at 37 °C while shaking at 300 rpm. A 4.5 μL aliquot was taken by pipetting on the wall of the tube (without mixing before). This sample corresponds to digest DNA. Step 3: Restriction enzyme inactivation and ligation of chimerical products. SDS (final concentration 0.2%) was added and samples were incubated at 37 °C for 30 min while shaking at 300 rpm. Samples were then diluted 4 times in 1X ligase buffer and Triton X-100, diluted in 1X ligase buffer, was added at 1% final concentration. Finally, samples were incubated at 37 °C for 2 h while shaking at 200 rpm. After a centrifugation of 1 min/2200 g/4 °C, 3.27 mL of supernatant were taken off such as 500 μL remain in the tube. On ice, 195 U of ligase (T4 DNA ligase, NEB M0202L) were carefully mixed and re-suspended with the pellet, then incubated overnight at 16 °C. After incubation this sample corresponds to 3C library. Step 4: Reversal of crosslinking. 3C libraries were 7 times diluted in 2X Proteinase K buffer (5 mM EDTA pH 8.0; 10 mM Tris-HCl pH 8.0; 0.5% SDS) and water. Undigested and digested samples were supplemented with 500 μL of 1xPK buffer. 100 μg or 20 μg of proteinase K (20 mg/mL) were mixed to the 3C libraries or to the undigested and digested DNA, respectively. All samples were incubated during 1 h at 50 °C and then during 4 h at 65 °C to reverse the crosslinking reaction. Step 5: DNA purification. 1 volume of Phenol:Chloroform:IAA was added to each sample (i.e., 4 mL to 3C library and 500 μL to the undigested and digested samples) and vigorously mixed. Following a centrifugation 10 min/16,100 g/RT, the supernatant aqueous phase was recovered in a new tube. 2 volumes of absolute ethanol and NaCl to a final concentration of 250 mM were added to each sample, mixed and let at −20° overnight to precipitate DNA. Following a centrifugation 20 min/16,100 g/4 °C, pellets were washed with 70% ethanol and finally re-suspended in water (150 μL for 3C libraries; 60 μL for the undigested and digested samples). Step 6: Qualitative PCR. After quantification at Nanodrop spectrophotometer, qualitative PCR (HiFi-Taq polymerase™ kit, Life Technologies, 11304-011) was carried out on 50 ng DNA of each samples by using the following primer pairs: Promoter-Gene, to detect genomic DNA (loading control); Promoter-Enhancer site, to detect the predicted interaction (3C products); Promoter-Enhancer nearby genomic region, as a negative control (non-interacting regions). Validated primers on
P2RY2 gene-enhancer interaction were used as a technique positive control [
41] and custom primers were used to study
DSCAM-AS1 promoter-enhancer interaction. Primers are reported in
Supplemental Table S6. PCR products were checked on 2% agarose-gel.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



