miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal



Abstract
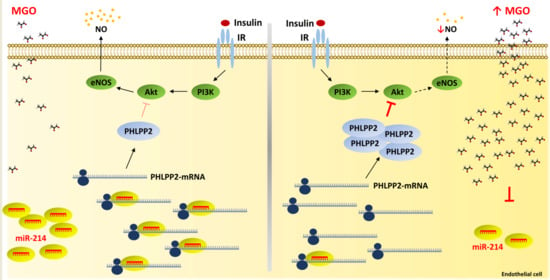
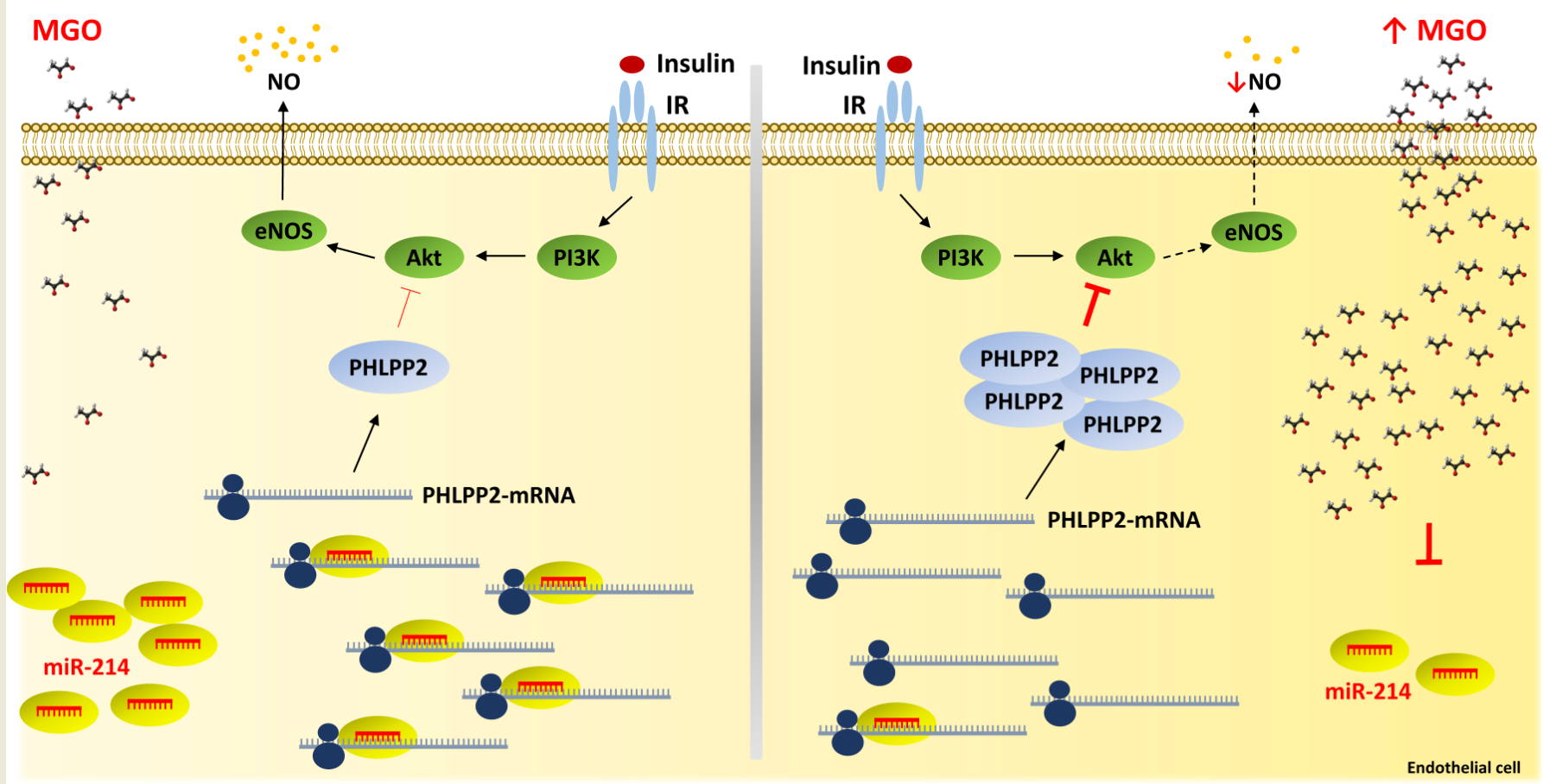
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
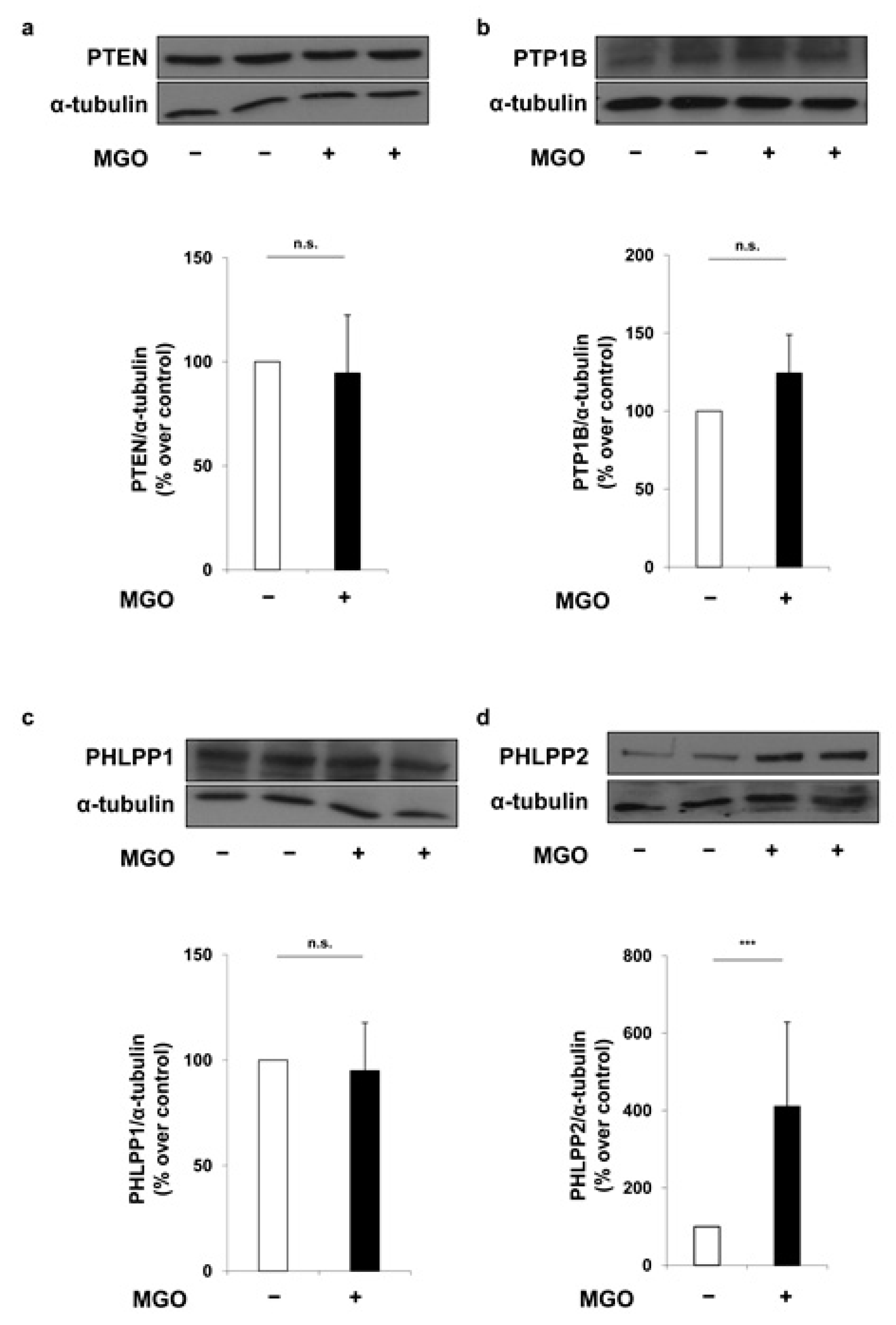
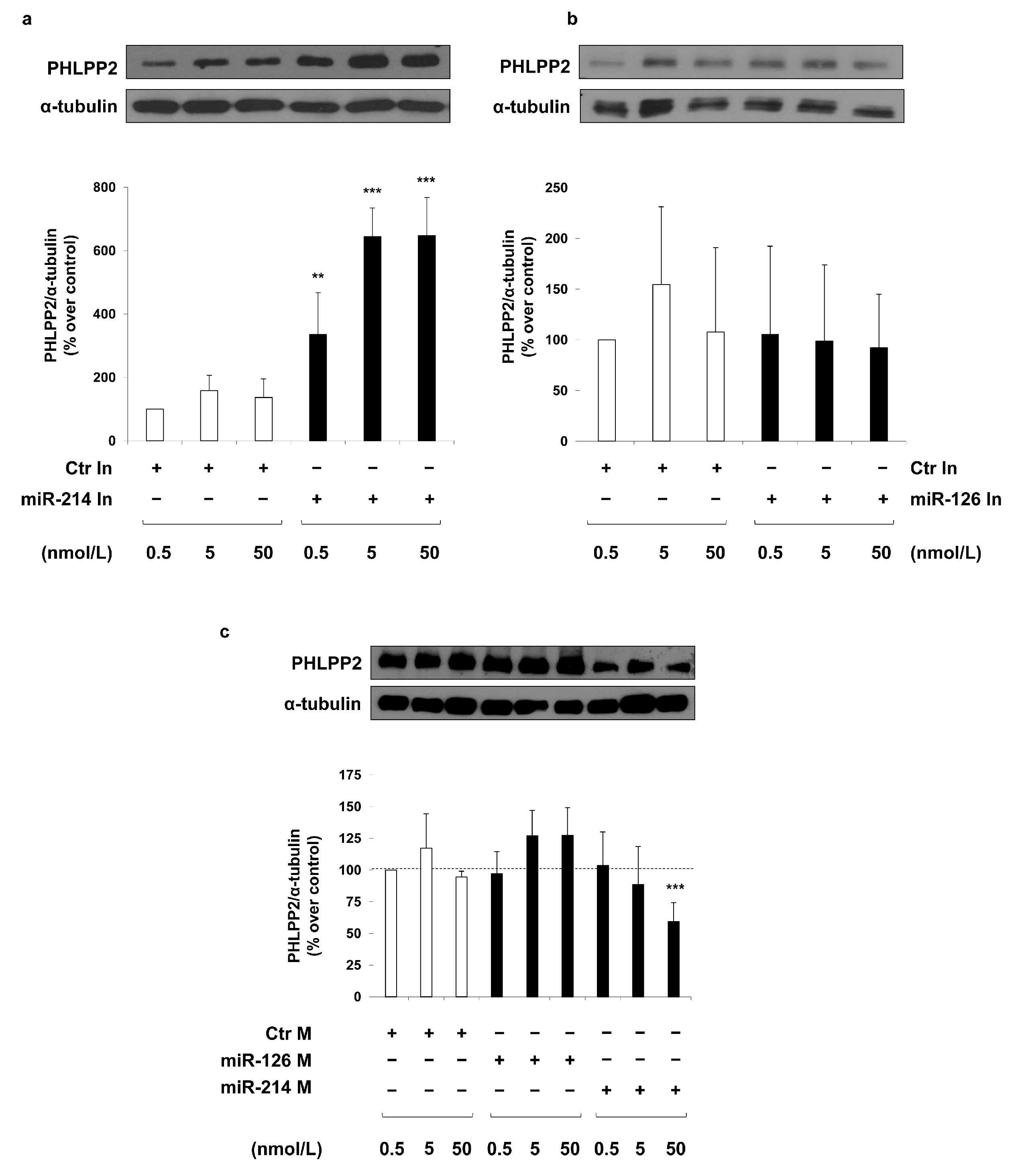
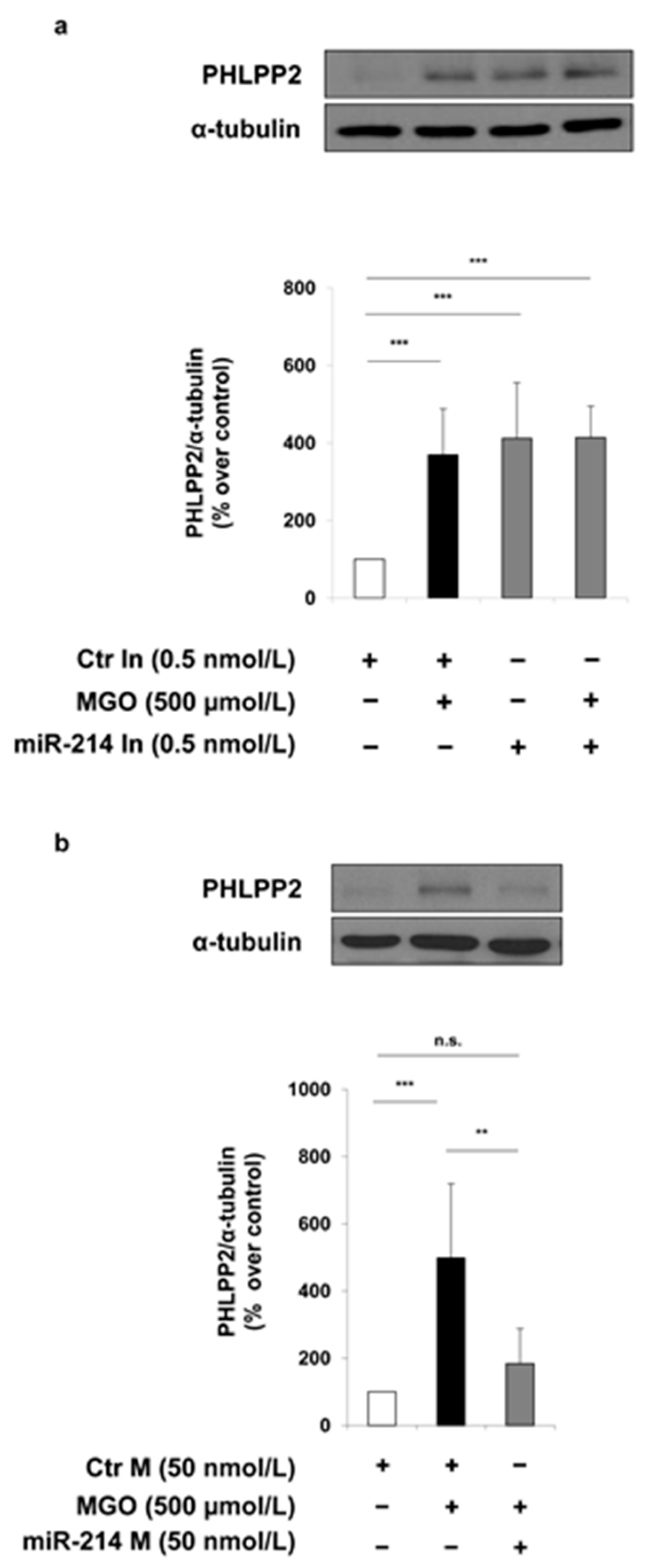
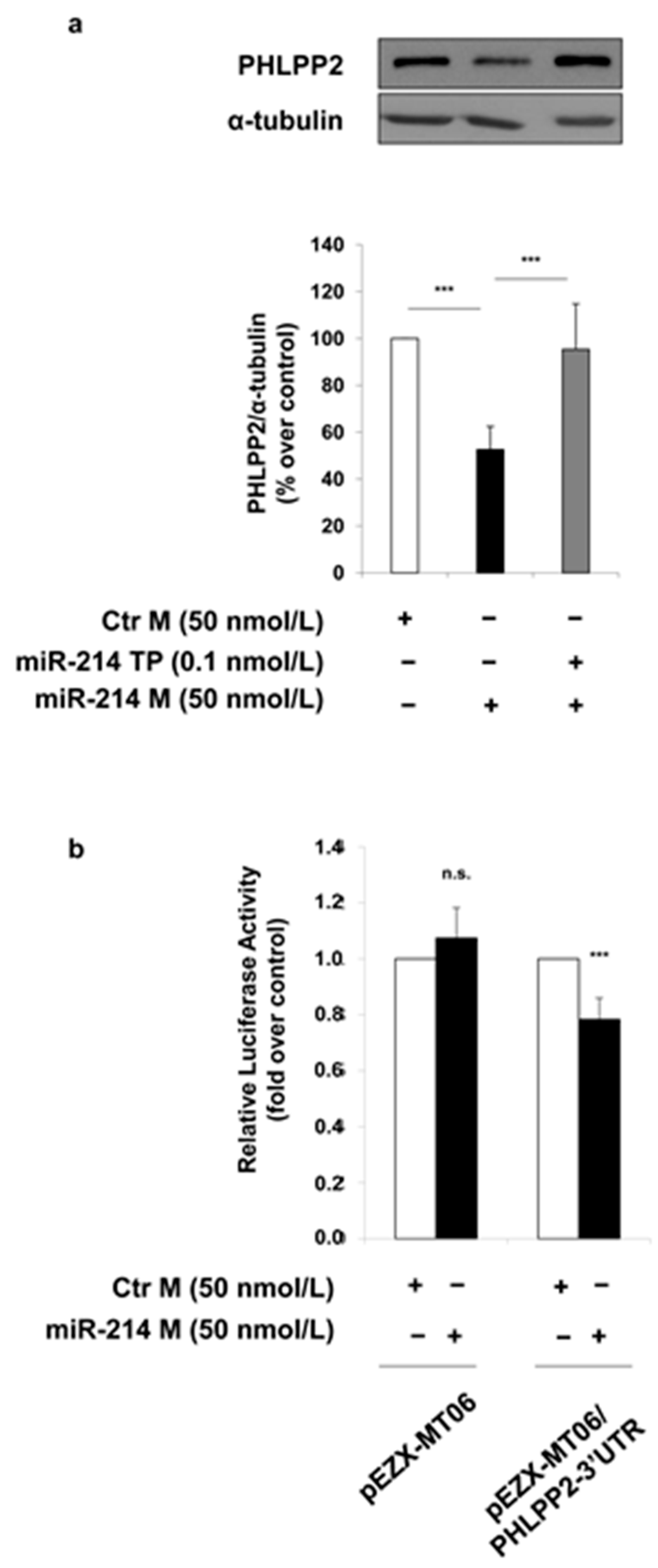
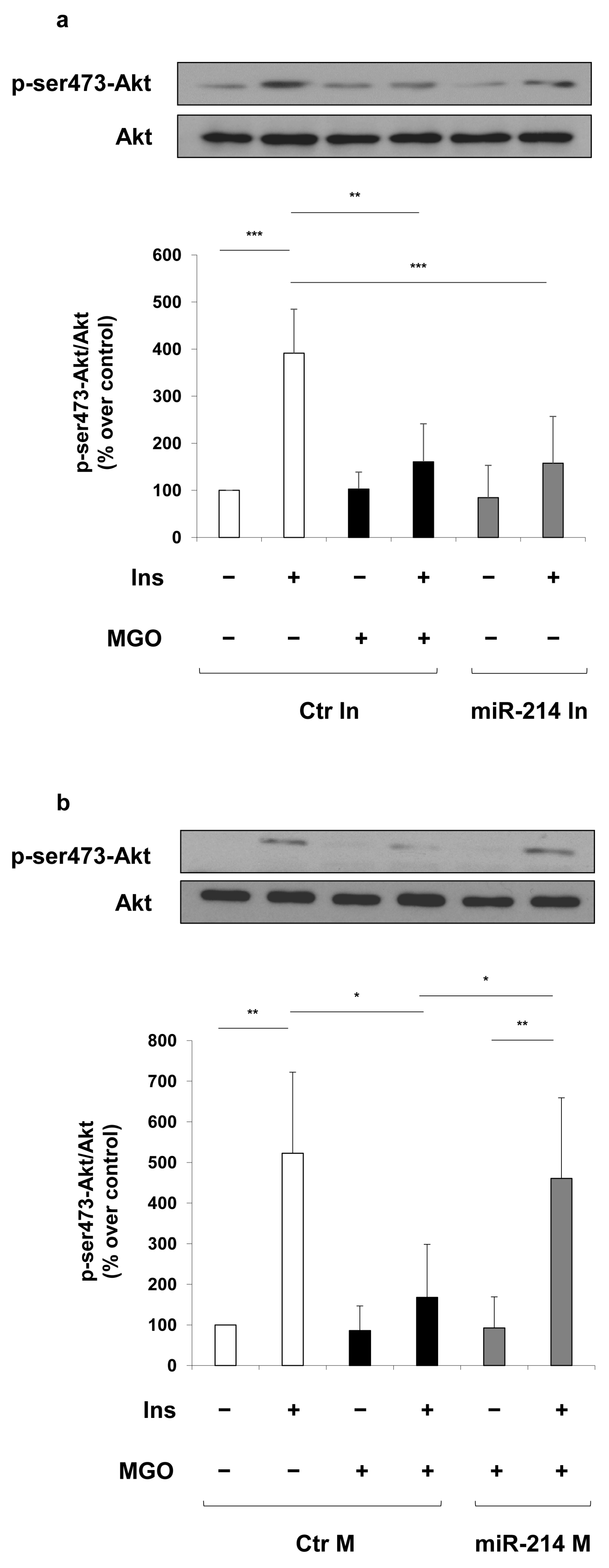
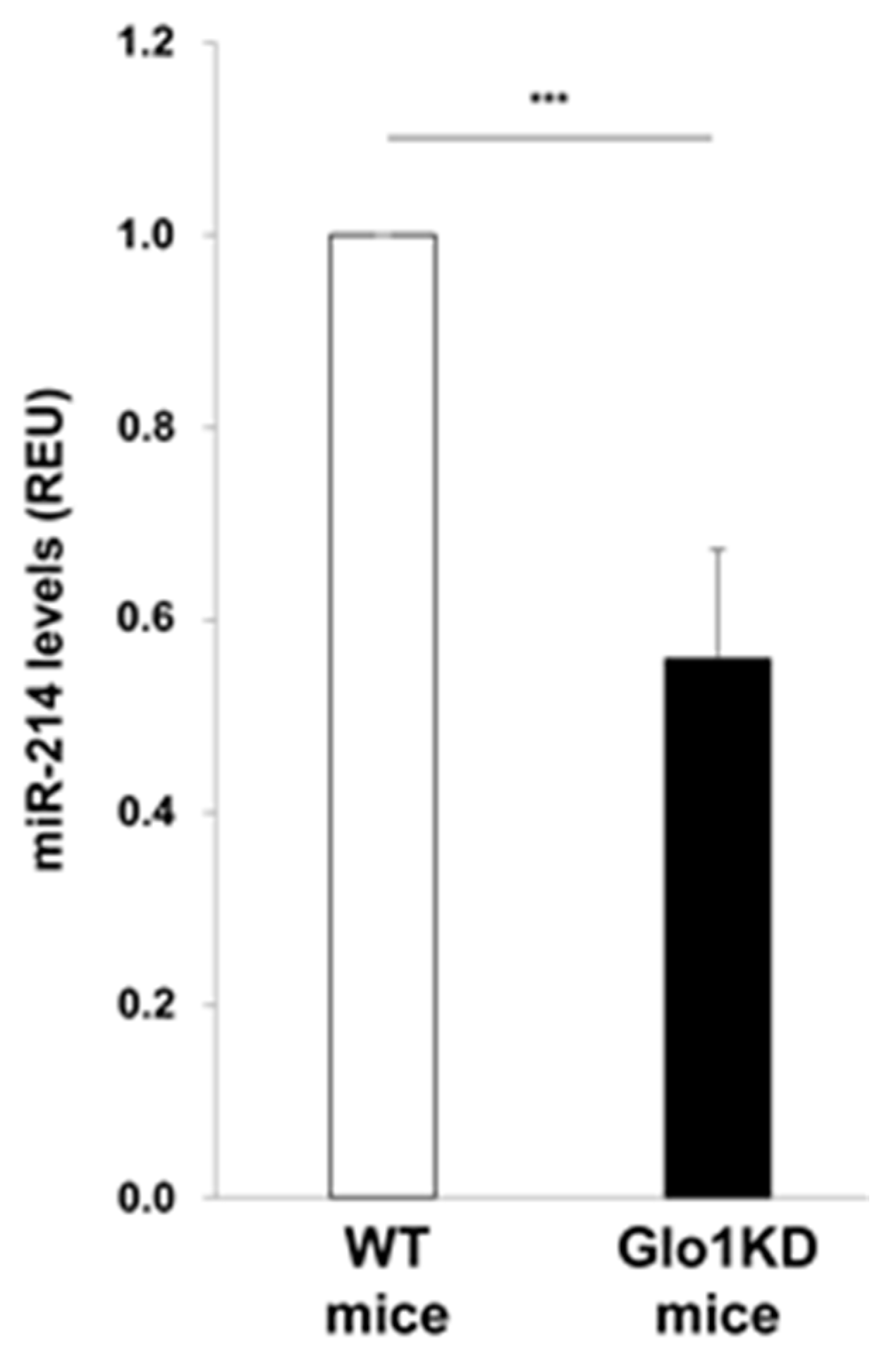
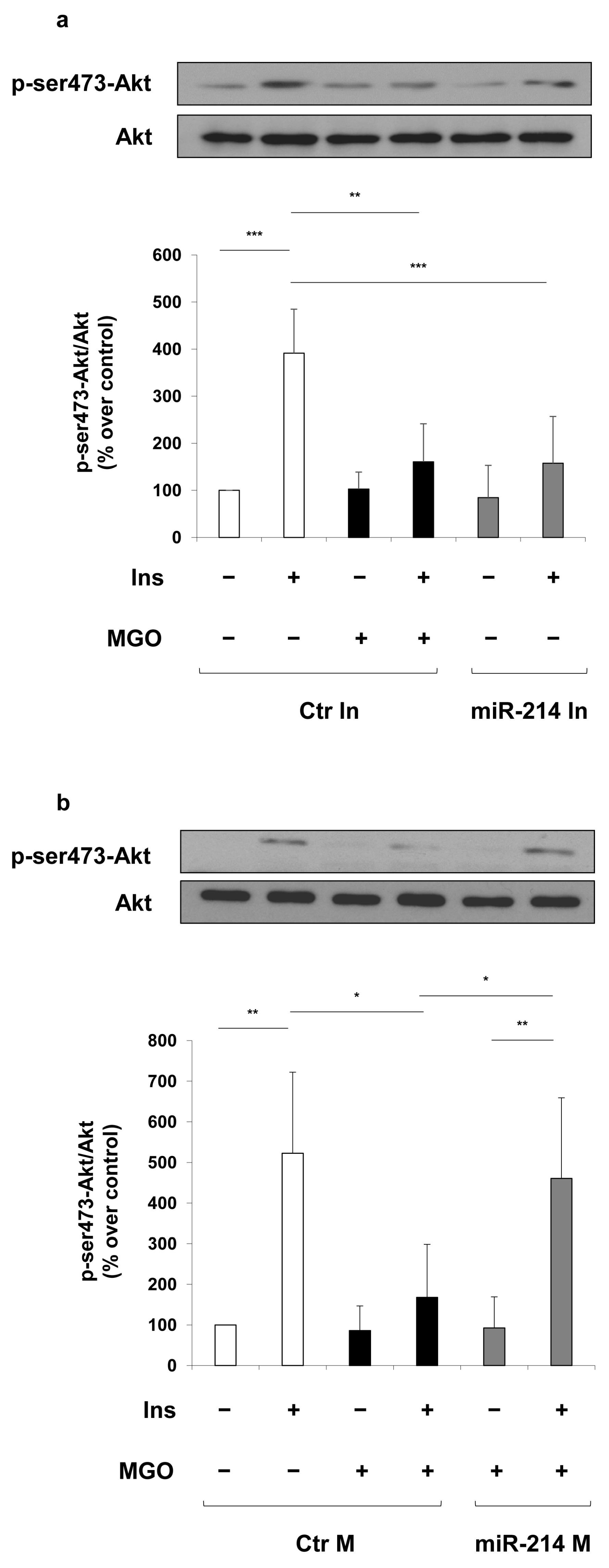
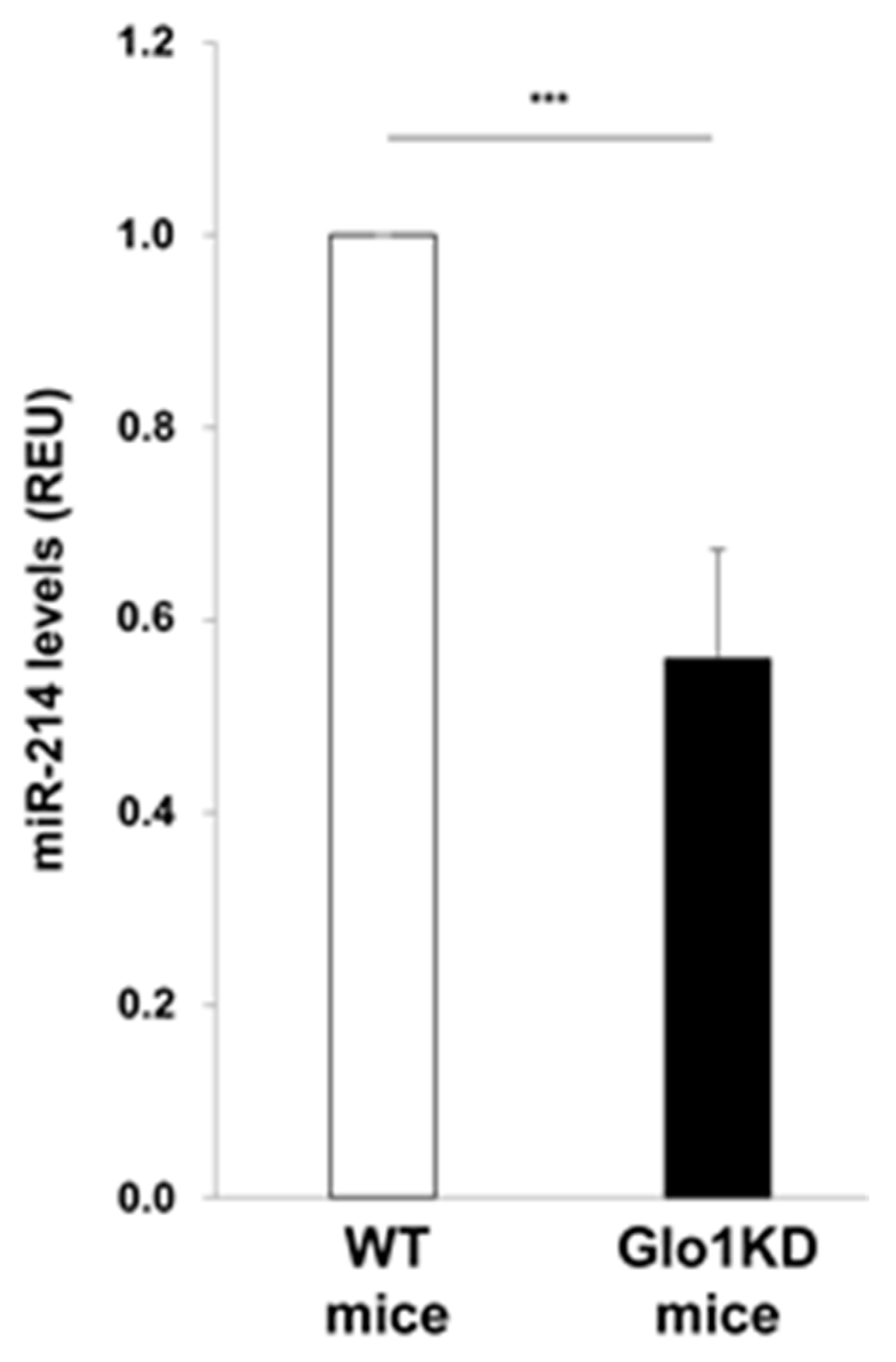
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. miRNA Target Prediction
4.4. miRNA Mimic and Inhibitor Transfection
4.5. Western Blot Analysis
4.6. miRNA Reverse Transcription, miScript PCR Array, and Real Time-PCR
- Mm_miR-214_2 miScript Primer Assay, MS00032571
- RNU6B_13 miScript Primer Assay, MS00014000
4.7. Target Site Inhibition Assays
4.8. Luciferase Reporter Assays
4.9. Statistic Procedures
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MGO | methylglyoxal |
| miRNAs | microRNAs |
| NO | nitric oxide |
| PTEN | phosphatase and tensin homolog |
| PTP1B | protein tyrosine phosphatase 1B |
| PHLPP1 | PH domain leucine-rich repeat protein phosphatase 1 |
| PHLPP2 | PH domain leucine-rich repeat protein phosphatase 2 |
| TP | target protector |
| T2DM | type 2 diabetes mellitus |
| UTR | untranslated region |
References
- Kahn, S.E. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Sun, X.; Shan, P.F. MicroRNAs and Cardiovascular Disease in Diabetes Mellitus. Biomed. Res. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.; Chen, H.; Barr, V.A.; Quon, M.J. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser (1179). J. Biol. Chem. 2001, 276, 30392–30398. [Google Scholar] [CrossRef] [PubMed]
- King, G.L.; Park, K.; Li, Q. Selective Insulin Resistance and the Development of Cardiovascular Diseases in Diabetes: The 2015 Edwin Bierman Award Lecture. Diabetes 2016, 65, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Kantharidis, P.; Wang, B.; Carew, R.M.; Lan, H.Y. Diabetes Complications: The MicroRNA Perspective. Diabetes 2011, 60, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Raciti, G.A.; Nigro, C.; Fiory, F.; D’Esposito, V.; Formisano, P.; Beguinot, F.; Miele, C. Circulating miRNAs as intercellular messengers, potential biomarkers and therapeutic targets for Type 2 diabetes. Epigenomics 2015, 7, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Zhu, L.; Gupta, N.; Chang, Y.; Fang, F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol. Endocrinol. 2007, 21, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Meng, X.; Che, H.; Shen, N.; Xiao, D.; Song, X.; Liang, M.; Fu, X.; Ju, J.; Li, Y.; et al. Regulation of insulin resistance by multiple miRNAs via targeting the GLUT4 signalling pathway. Cell. Physiol. Biochem. 2016, 38, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Lin, J.; Zhang, Y.; Kang, S.; Belkin, N.; Wara, A.K.; Icli, B.; Hamburg, N.M.; Li, D.; Feinberg, M.W. MicroRNA-181b improves glucose homeostasis and insulin sensitivity by regulating endothelial function in white adipose tissue. Circ. Res. 2016, 118, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, G.; Lakshmanan, A.P.; Samuel, S.M.; Triggle, C.R.; Ding, H. Molecular interplay between microRNA-34a and sirtuin1 in hyperglycemia-mediated impaired angiogenesis in endothelial cells: Effects of metformins. J. Pharmacol. Exp. Ther. 2016, 356, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Goumenos, A.; Adamopoulos, C.; Papavassiliou, A.G. AGE/RAGE signalling regulation by miRNAs: Associations with diabetic complications and therapeutic potential. Int. J. Biochem. Cell Biol. 2015, 60, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Leone, A.; Raciti, G.A.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-Glyoxalase 1 balance: The root of vascular damage. Int. J. Mol. Sci. 2017, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, N.; Tian, D. MiR-30b is involved in methylglyoxal-induced epithelial-mesenchymal transition of peritoneal mesothelial cells in rats. Cell. Mol. Biol. Lett. 2014, 19, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Wu, Y.; Jin, X.; Jiang, C. The SUR2B subunit of rat vascular KATP channel is targeted by miR-9a-3p induced by prolonged exposure to methylglyoxal. Am. J. Physiol. Cell Physiol. 2015, 308, C139–C145. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Raciti, G.A.; Leone, A.; Fleming, T.H.; Longo, M.; Prevenzano, I.; Fiory, F.; Mirra, P.; D’Esposito, V.; Ulianich, L.; et al. Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia 2014, 57, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Nigro, C.; Prevenzano, I.; Procopio, T.; Leone, A.; Raciti, G.A.; Andreozzi, F.; Longo, M.; Fiory, F.; Beguinot, F.; et al. The role of miR-190a in methylglyoxal-induced insulin resistance in endothelial cells. Biochim. Biophys. Acta 2017, 1863, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in non-diabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, N.W.; Hansen, A.J.; Sams, A. The endothelial border to health: Mechanistic evidence of the hyperglycemic culprit of inflammatory disease acceleration. IUBMB Life 2017, 69, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.P.; Rawal, K.; Bagchi, A.K.; Akolkar, G.; Bernardes, N.; Dias Dda, S.; Gupta, S.; Singal, P.K. Insulin resistance: An additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail. Rev. 2016, 21, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Putta, S.; Kato, M. MicroRNAs and diabetic complications. J. Cardiovasc. Transl. Res. 2012, 5, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, X.; Icli, B.; Feinberg, M.W. Emerging Roles for MicroRNAs in Diabetic Microvascular Disease: Novel Targets for Therapy. Endocr. Rev. 2017, 38, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kim, G.D.; Park, H.R.; Park, Y.S. Comparative mRNA and microRNA expression profiling of methylglyoxal exposed human endothelial cells. BioChip J. 2013, 7, 143–150. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. The role of miR-126 in embryonic angiogenesis, adult vascular homeostasis, and vascular repair and its alterations in atherosclerotic disease. J. Mol. Cell. Cardiol. 2016, 97, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Rippe, C.; Blimline, M.; Magerko, K.A.; Lawson, B.R.; LaRocca, T.J.; Donato, A.J.; Seals, D.R. MicroRNA changes in human arterial endothelial cells with senescence: Relation to apoptosis, eNOS and inflammation. Exp. Gerontol. 2012, 47, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yang, C.J.; Xu, X.; Cao, J.N.; Feng, Q.T.; Yang, J. MiR-214 regulates the pathogenesis of patients with coronary artery disease by targeting VEGF. Mol. Cell. Biochem. 2015, 402, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Van Mil, A.; Grundmann, S.; Goumans, M.J.; Lei, Z.; Oerlemans, M.I.; Jaksani, S.; Doevendans, P.A.; Sluijter, J.P. MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc. Res. 2012, 93, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Van Balkom, B.W.; de Jong, O.G.; Smits, M.; Brummelman, J.; den Ouden, K.; de Bree, P.M.; van Eijndhoven, M.A.; Pegtel, D.M.; Stoorvogel, W.; Würdinger, T.; et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013, 121, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Yang, L.; Gong, W.; Chaugai, S.; Wang, F.; Chen, C.; Wang, P.; Zou, M.H.; Wang, D.W. MicroRNA-214 Is Upregulated in Heart Failure Patients and Suppresses XBP1-Mediated Endothelial Cells Angiogenesis. J. Cell. Physiol. 2015, 230, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, Y.Y.; Lin, Y.F.; Chen, Y.T.; Yang, L.; Wang, H.J.; Ma, D. The cell growth suppressor, miR-126, targets IRS-1. Biochem. Biophys. Res. Commun. 2008, 377, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.S.; Park, S.Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343. [Google Scholar] [CrossRef]
- Wang, X.; Shen, E.; Wang, Y.; Li, J.; Cheng, D.; Chen, Y.; Gui, D.; Wang, N. Cross talk between miR-214 and PTEN attenuates glomerular hypertrophy under diabetic conditions. Sci. Rep. 2016, 6, 31506. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V. MicroRNA expression profiling in human ovarian cancer: mir-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, S. miR-214 promotes radioresistance in human ovarian cancer cells by targeting PTEN. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, W.; Zhang, H.; Liu, T.; Zhao, L. miR-214 targets the PTEN-mediated PI3K/Akt signaling pathway and regulates cell proliferation and apoptosis in ovarian cancer. Oncol. Lett. 2017, 14, 5711–5718. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.S.; Yang, X.H.; Wang, X.D.; Wang, Y.L.; Zhou, B.; Song, Z.S. MiR-214 regulate gastric cancer cell proliferation, migration and invasion by targeting PTEN. Cancer Cell Int. 2013, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Xin, R.; Bai, F.; Feng, Y.; Jiu, M.; Liu, X.; Bai, F.; Nie, Y.; Fan, D. MicroRNA-214 promotes peritoneal metastasis through regulating PTEN negatively in gastric cancer. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Honardoost, M.; Soleimani, M.; Arefian, E.; Sarookhani, M.R. Expression Change of miR-214 and miR-135 during Muscle Differentiation. Cell J. 2015, 17, 461–470. [Google Scholar] [PubMed]
- Wang, X.Y.; Bergdahl, K.; Heijbel, A.; Liljebris, C.; Bleasdale, J.E. Analysis of in vitro interactions of protein tyrosine phosphatase 1B with insulin receptors. Mol. Cell. Endocrinol. 2001, 173, 109–120. [Google Scholar] [CrossRef]
- Conche, C.; Sauer, K. Uncovering the PI3Ksome: Phosphoinositide 3-kinases and counteracting PTEN form a signalingcomplex with intrinsic regulatory properties. Mol. Cell. Biol. 2014, 34, 3356–3358. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C.; Trotman, L.C. Turning off Akt: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 537–558. [Google Scholar] [CrossRef] [PubMed]
- Grzechnik, A.T.; Newton, A.C. PHLPPing through history: A decade in the life of PHLPP phosphatases. Biochem. Soc. Trans. 2016, 44, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Pandey, V.K.; Kakkar, P. PHLPP: A putative cellular target during insulin resistance and type 2 diabetes. J. Endocrinol. 2017, 233, R185–R198. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.P.; Goodall, G.J.; Liu, B. Understanding principles of miRNA target recognition and function through integrated biological and bioinformatics approaches. Wiley Interdiscip. Rev. RNA 2014, 5, 361–379. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigro, C.; Mirra, P.; Prevenzano, I.; Leone, A.; Fiory, F.; Longo, M.; Cabaro, S.; Oriente, F.; Beguinot, F.; Miele, C. miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal. Int. J. Mol. Sci. 2018, 19, 522. https://doi.org/10.3390/ijms19020522
Nigro C, Mirra P, Prevenzano I, Leone A, Fiory F, Longo M, Cabaro S, Oriente F, Beguinot F, Miele C. miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal. International Journal of Molecular Sciences. 2018; 19(2):522. https://doi.org/10.3390/ijms19020522
Chicago/Turabian StyleNigro, Cecilia, Paola Mirra, Immacolata Prevenzano, Alessia Leone, Francesca Fiory, Michele Longo, Serena Cabaro, Francesco Oriente, Francesco Beguinot, and Claudia Miele. 2018. "miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal" International Journal of Molecular Sciences 19, no. 2: 522. https://doi.org/10.3390/ijms19020522
APA StyleNigro, C., Mirra, P., Prevenzano, I., Leone, A., Fiory, F., Longo, M., Cabaro, S., Oriente, F., Beguinot, F., & Miele, C. (2018). miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal. International Journal of Molecular Sciences, 19(2), 522. https://doi.org/10.3390/ijms19020522