Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure

Abstract
1. Introduction
2. Results
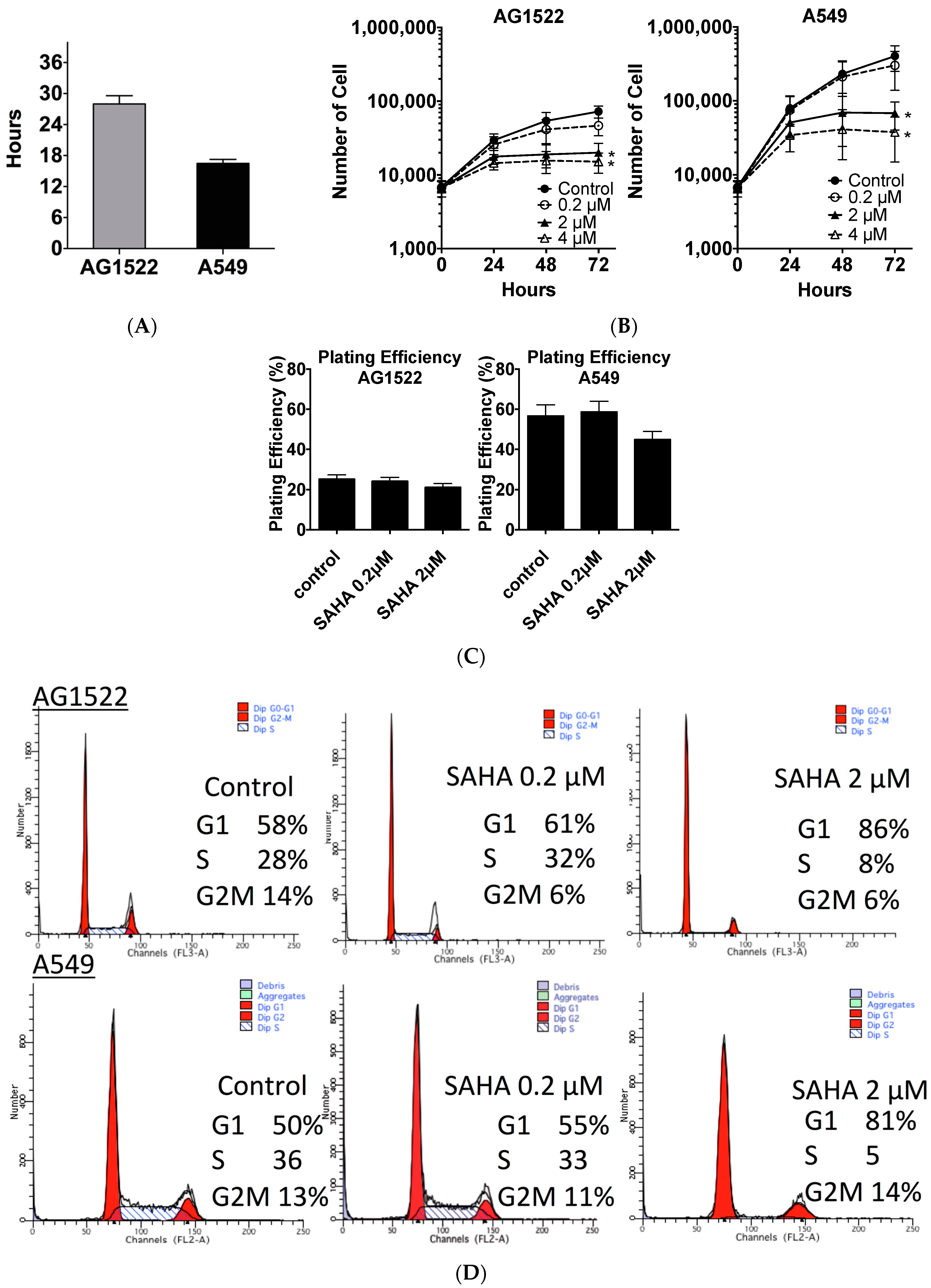
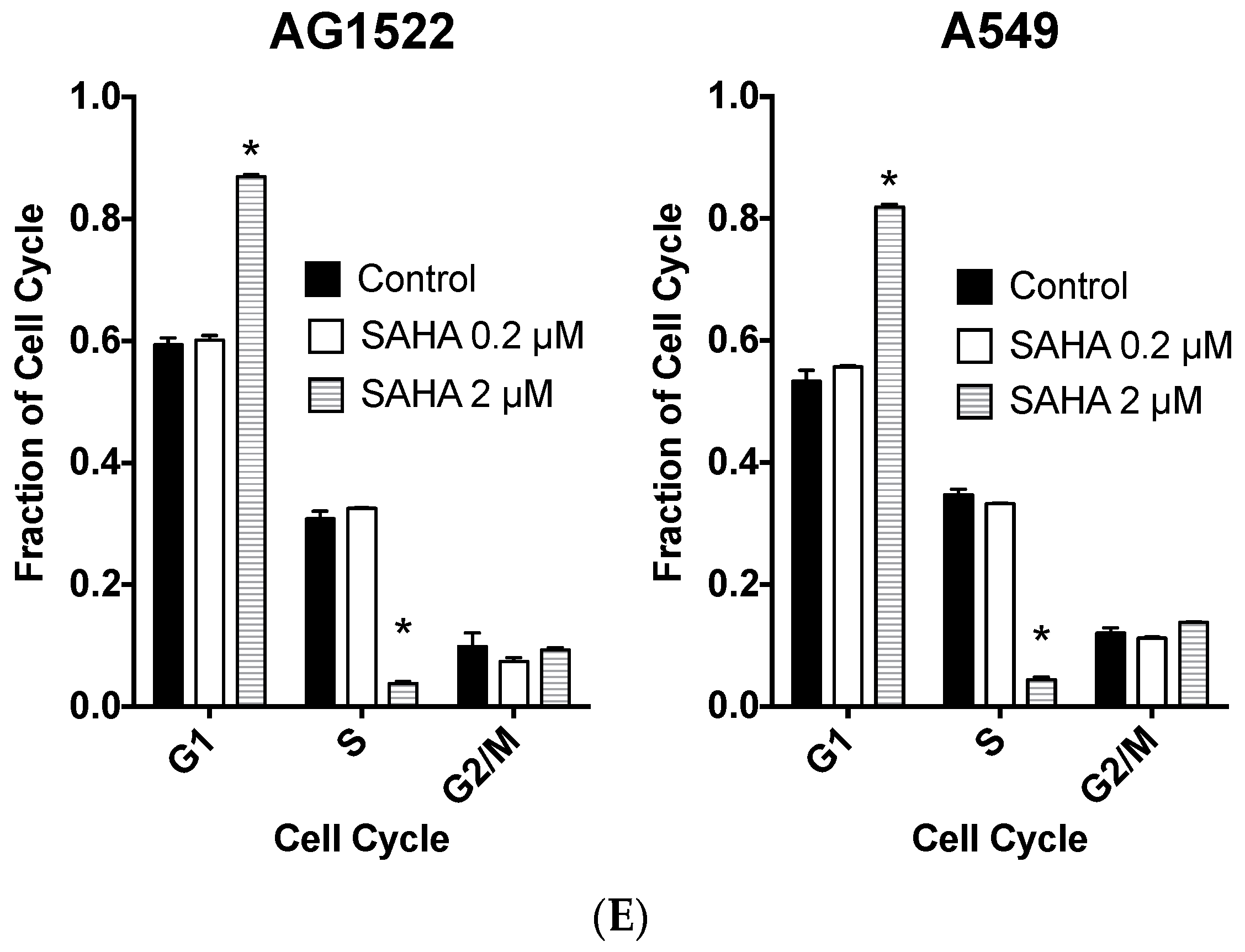
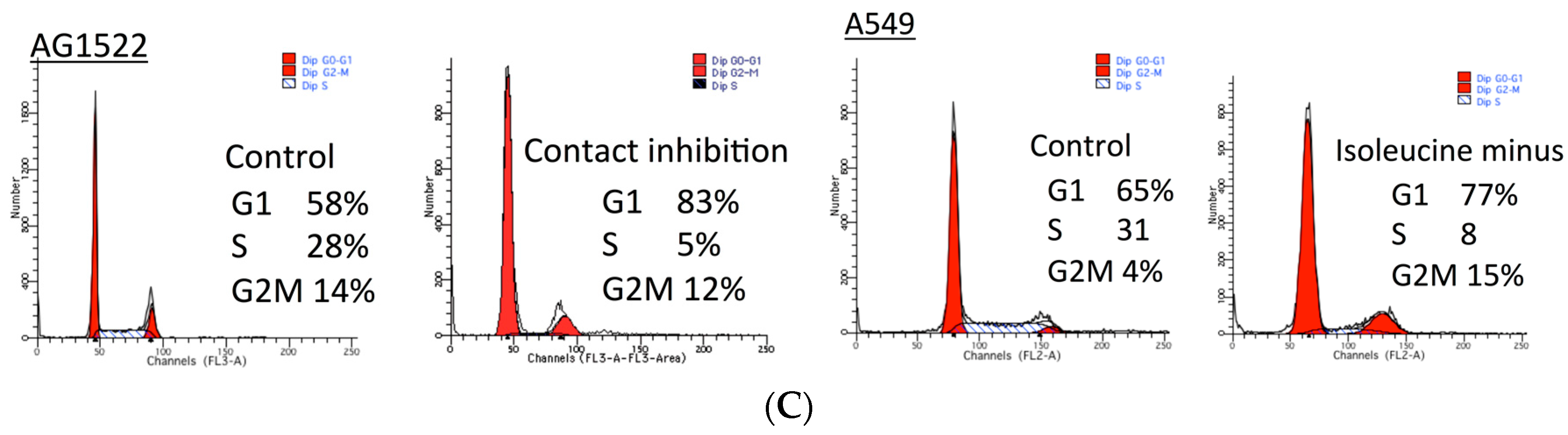
2.1. Cellular Toxicity and Cell Cycle Distribution from SAHA Treatment
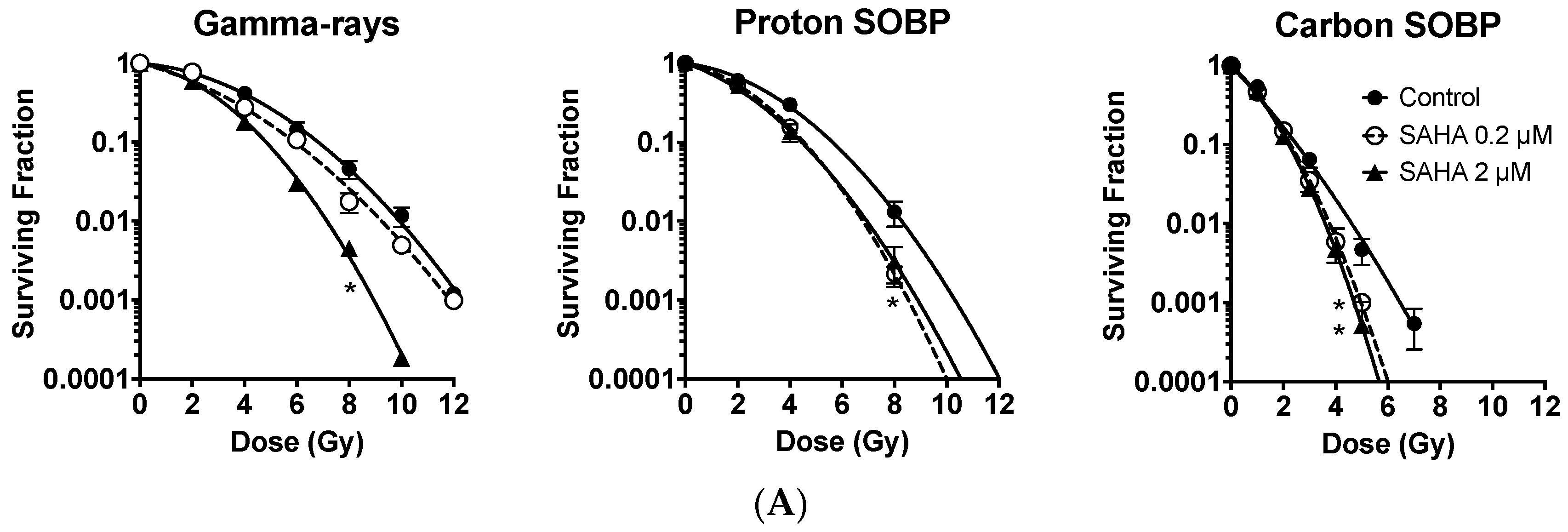
2.2. Radiosensitization of Exponentially Growing Cancer Cells Exposed to γ-Rays, Protons and Clinical Grade Carbon Ions
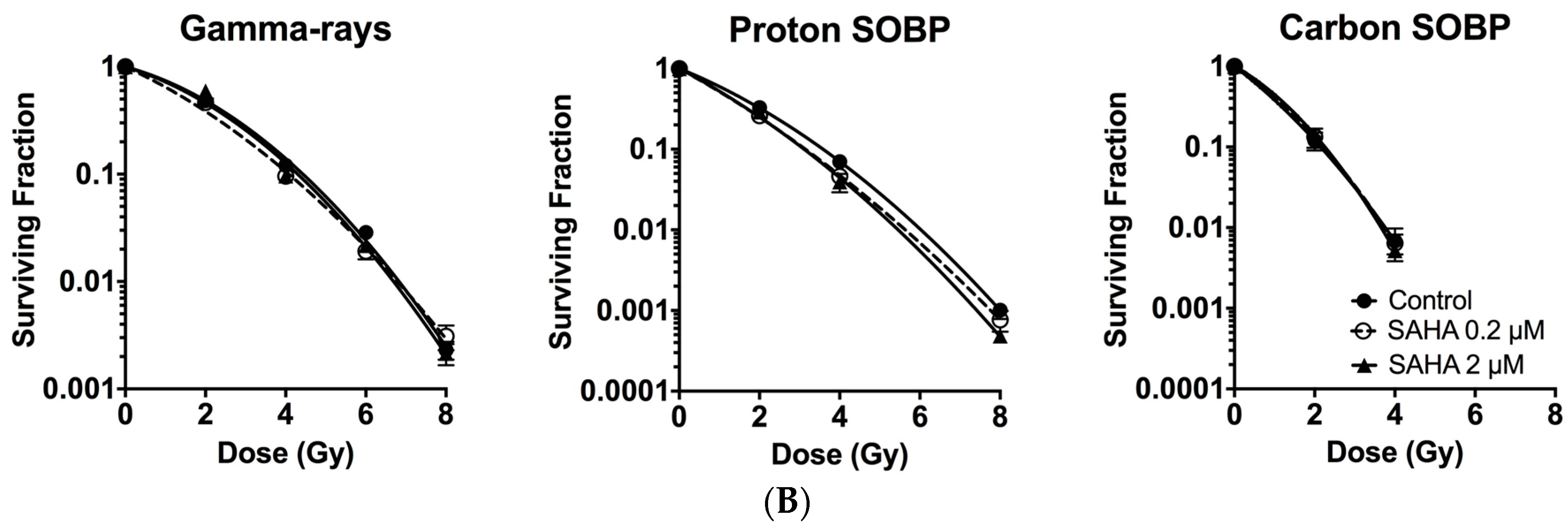
2.3. Minimum Radiosensitization Effect of SAHA for Exponentially Growing Normal Cells
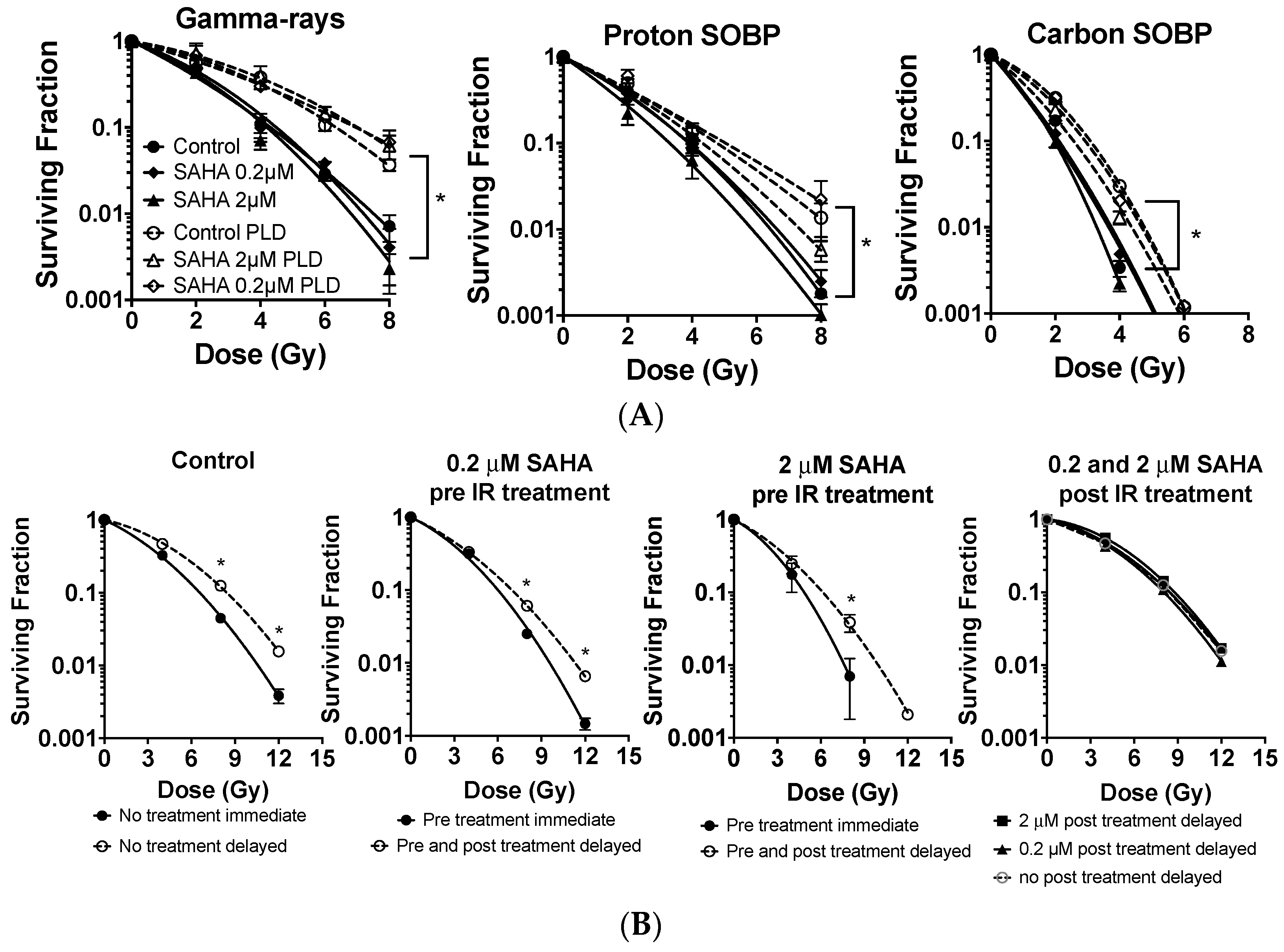
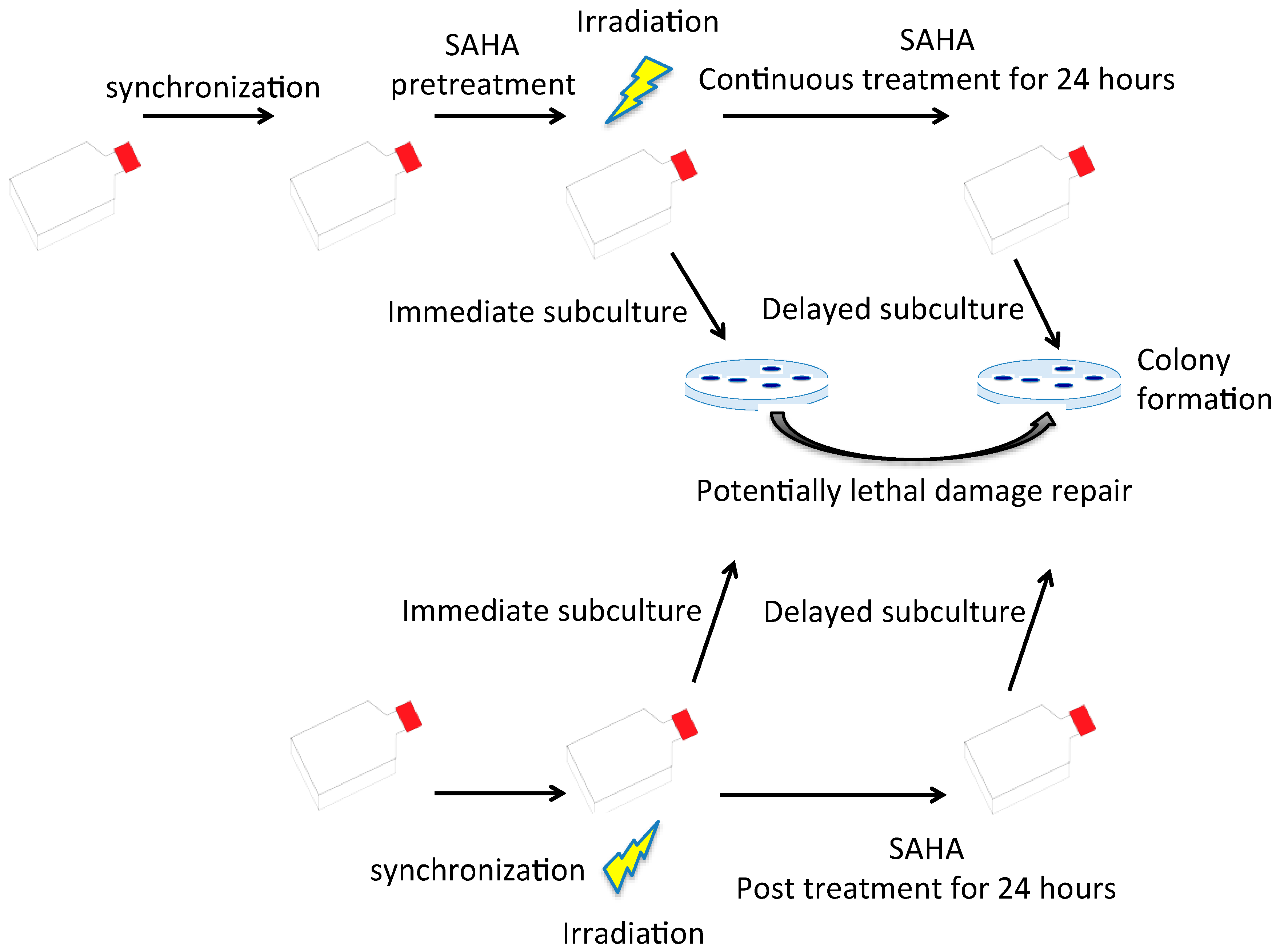
2.4. G0/G1 Phase Cells and Potentially Lethal Damage Repair with Immediate or Delayed Subculture
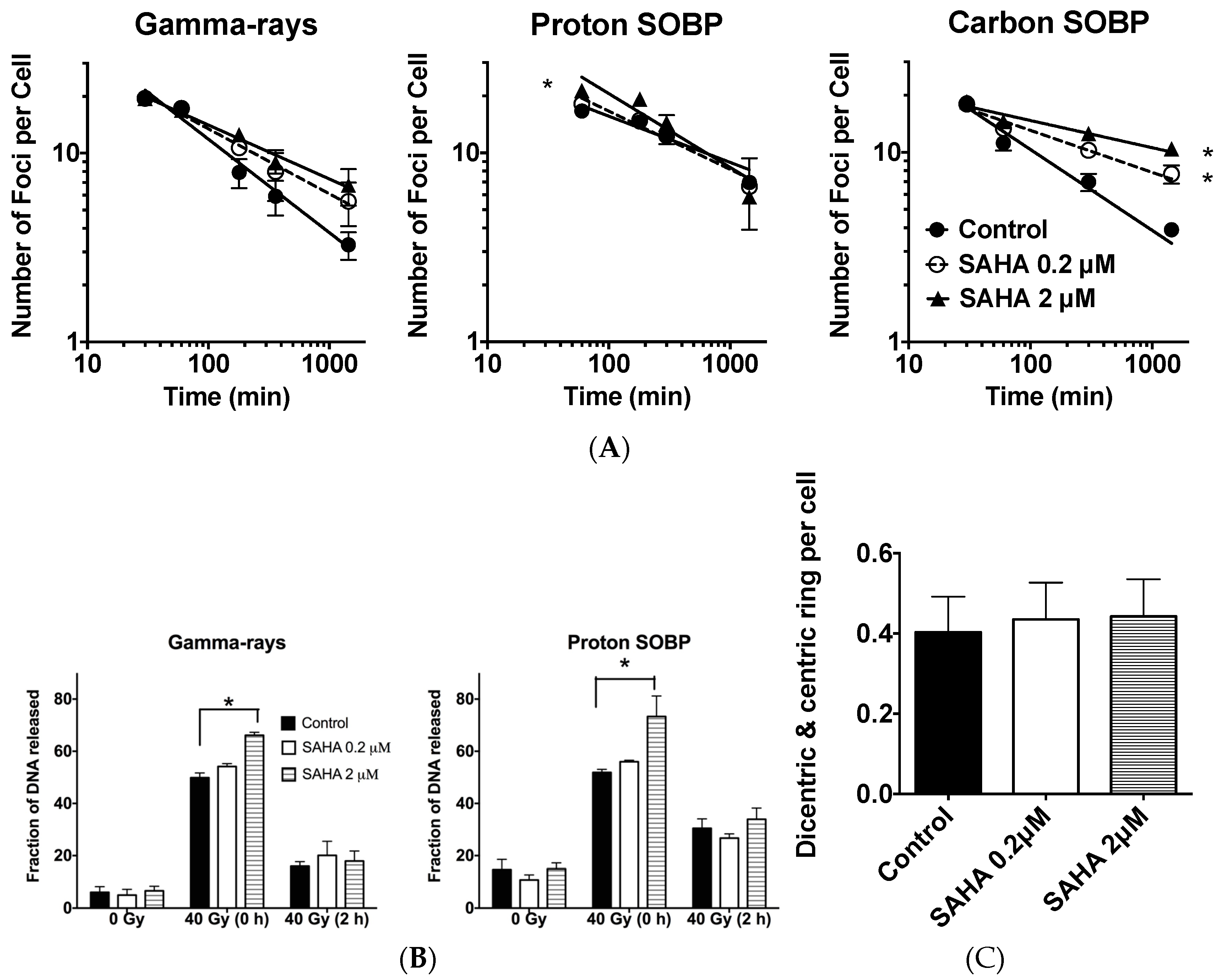
2.5. DSB (Double Starand Break) Repair
2.6. Chromosomal Aberrations
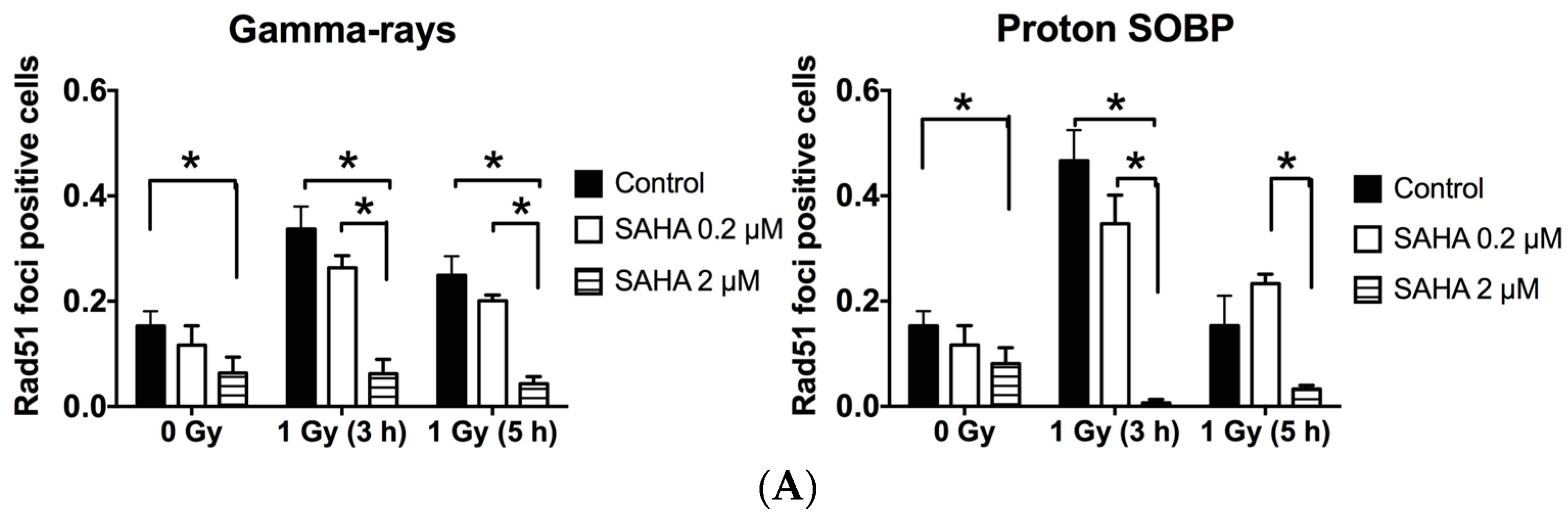
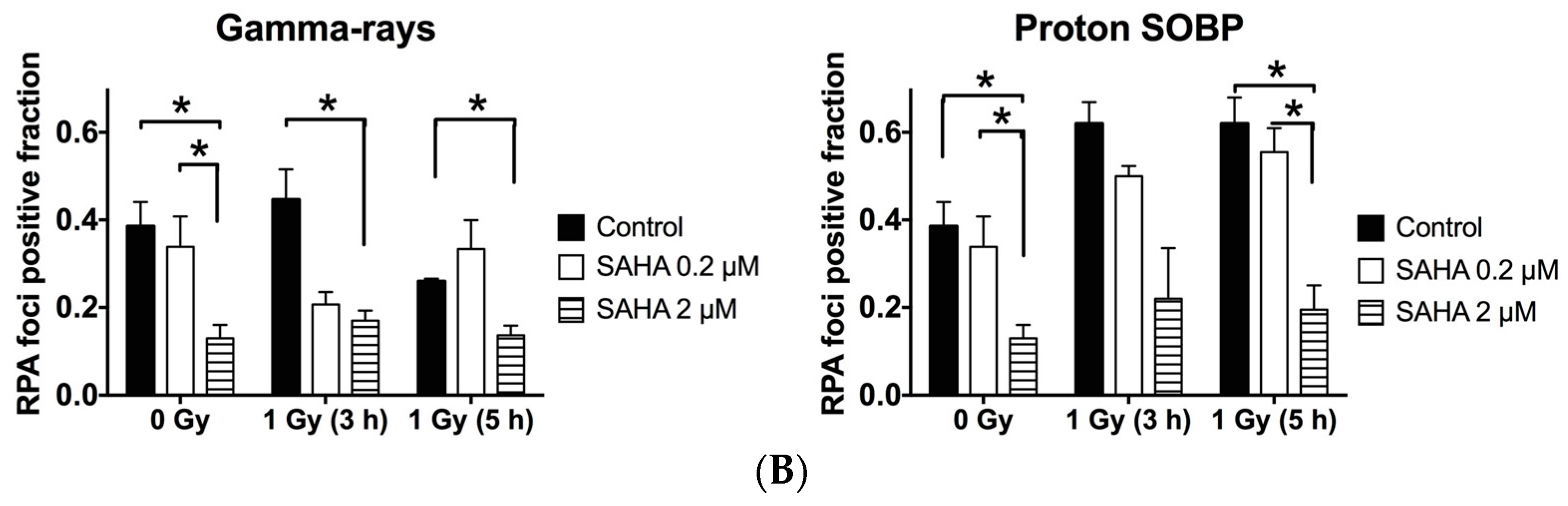
2.7. Rad51 and RPA (Replication Protein A) Focus Formation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Cycle Synchronization
4.3. Irradiation
4.4. Cellular Toxicity and Cell Cycle Analysis
4.5. Cell Survival Curves and RBE Analysis
4.6. Potentially Lethal Damage Repair Analysis with Immediate or Delayed Subculture
4.7. Immunostaining and DNA Damage Response Analysis
4.8. Chromosome Aberration Analysis
4.9. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SAHA | Suberoylanilide hydroxamic acid |
| HDACi | Histone deacetylase inhibitor |
| LET | Linear energy transfer |
| RBE | Relative biological effectiveness |
| SER | Sensitization Enhancement Ratio |
| D10 | Dose to achieve 10% cell survival |
| PMRC | Proton Medical Research Center |
| HIMAC | Heavy ion medical accelerator |
| SOBP | Spread out Bragg peak |
| ANOVA | Analysis of valiance |
References
- Fujii, Y.; Genet, M.D.; Roybal, E.J.; Kubota, N.; Okayasu, R.; Miyagawa, K.; Fujimori, A.; Kato, T.A. Comparison of the bromodeoxyuridine-mediated sensitization effects between low-let and high-let ionizing radiation on DNA double-strand breaks. Oncol. Rep. 2013, 29, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.W.; Loaiza-Bonilla, A.; Juckett, M.; DiPersio, J.F.; Roy, V.; Slack, J.; Wu, W.; Laumann, K.; Espinoza-Delgado, I.; Gore, S.D.; et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica 2009, 94, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, S.M.; White, L.A.; Kolesar, J.M. Vorinostat: A novel therapy for the treatment of cutaneous t-cell lymphoma. Am. J. Health-Syst. Pharm. 2010, 67, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Adachi, M.; Zou, H.; Hareyama, M.; Imai, K.; Shinomura, Y. Histone deacetylase inhibitors enhance phosphorylation of histone h2ax after ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Kvarnbrink, S.; Holmlund, C.; Wibom, C.; Bergenheim, T.; Henriksson, R.; Hedman, H. Synergistic killing of glioblastoma stem-like cells by bortezomib and hdac inhibitors. Anticancer Res. 2012, 32, 2407–2413. [Google Scholar] [PubMed]
- Di Bernardo, G.; Alessio, N.; Dell’Aversana, C.; Casale, F.; Teti, D.; Cipollaro, M.; Altucci, L.; Galderisi, U. Impact of histone deacetylase inhibitors saha and ms-275 on DNA repair pathways in human mesenchymal stem cells. J. Cell. Physiol. 2010, 225, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Frame, F.M.; Pellacani, D.; Collins, A.T.; Simms, M.S.; Mann, V.M.; Jones, G.D.; Meuth, M.; Bristow, R.G.; Maitland, N.J. Hdac inhibitor confers radiosensitivity to prostate stem-like cells. Br. J. Cancer 2013, 109, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; Harikrishnan, K.N.; El-Osta, A. Disparity of histone deacetylase inhibition on repair of radiation-induced DNA damage on euchromatin and constitutive heterochromatin compartments. Oncogene 2007, 26, 3963–3971. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wong, P.; Radany, E.H.; Stark, J.M.; Laulier, C.; Wong, J.Y. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of rad51 protein and inhibition of homology-directed repair in multiple myeloma. Mol. Cancer Res. 2012, 10, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Radany, E.H.; Wong, P.; Ma, S.; Wu, K.; Wang, B.; Wong, J.Y. Suberoylanilide hydroxamic acid induces hypersensitivity to radiation therapy in acute myelogenous leukemia cells expressing constitutively active flt3 mutants. PLoS ONE 2013, 8, e84515. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. Hdac inhibitor pci-24781 decreases rad51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [PubMed]
- Ladd, B.; Ackroyd, J.J.; Hicks, J.K.; Canman, C.E.; Flanagan, S.A.; Shewach, D.S. Inhibition of homologous recombination with vorinostat synergistically enhances ganciclovir cytotoxicity. DNA Repair 2013, 12, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Yang, X.; Sottero, T.L.; Gragg, A.; Prasad, G.; Polley, M.Y.; Weiss, W.A.; Matthay, K.K.; Davidoff, A.M.; DuBois, S.G.; et al. Cooperation of the hdac inhibitor vorinostat and radiation in metastatic neuroblastoma: Efficacy and underlying mechanisms. Cancer Lett. 2011, 306, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Hada, M.; Opipari, A.W., Jr.; Castle, V.P.; Kwok, R.P. Creb-binding protein regulates ku70 acetylation in response to ionization radiation in neuroblastoma. Mol. Cancer Res. 2013, 11, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Oertel, S.; Thiemann, M.; Richter, K.; Weber, K.J.; Huber, P.E.; Perez, R.L.; Brons, S.; Bischof, M.; Kulozik, A.E.; Ehemann, V.; et al. Combination of suberoylanilide hydroxamic acid with heavy ion therapy shows promising effects in infantile sarcoma cell lines. Radiat. Oncol. 2011, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Jeynes, J.C.; Merchant, M.J.; Wera, A.C.; Barry, M.A.; Kirkby, K.J.; Suzuki, M. Radiosensitization of glioblastoma cells using a histone deacetylase inhibitor (saha) comparing carbon ions with X-rays. Int. J. Radiat. Biol. 2015, 91, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Yamada, S.; Hoshino, I.; Murakami, K.; Akutsu, Y.; Sakata, H.; Nishimori, T.; Usui, A.; Miyazawa, Y.; Kamada, T.; et al. Effects of carbon-ion radiotherapy combined with a novel histone deacetylase inhibitor, cyclic hydroxamic-acid-containing peptide 31 in human esophageal squamous cell carcinoma. Anticancer Res. 2009, 29, 4433–4438. [Google Scholar] [PubMed]
- Saito, K.; Funayama, T.; Yokota, Y.; Murakami, T.; Kobayashi, Y. Histone deacetylase inhibitors sensitize murine b16f10 melanoma cells to carbon ion irradiation by inducing g1 phase arrest. Biol. Pharm. Bull. 2017, 40, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Wang, A.J.; Yao, L.F. Antitumor histone deacetylase inhibitors suppress cutaneous radiation syndrome: Implications for increasing therapeutic gain in cancer radiotherapy. Mol. Cancer Ther. 2004, 3, 317–325. [Google Scholar] [PubMed]
- Brown, S.L.; Kolozsvary, A.; Liu, J.; Ryu, S.; Kim, J.H. Histone deacetylase inhibitors protect against and mitigate the lethality of total-body irradiation in mice. Radiat. Res. 2008, 169, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Blattmann, C.; Oertel, S.; Thiemann, M.; Weber, K.J.; Schmezer, P.; Zelezny, O.; Lopez Perez, R.; Kulozik, A.E.; Debus, J.; Ehemann, V. Suberoylanilide hydroxamic acid affects γh2ax expression in osteosarcoma, atypical teratoid rhabdoid tumor and normal tissue cell lines after irradiation. Strahlenther. Onkol. 2012, 188, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Blattmann, C.; Oertel, S.; Ehemann, V.; Thiemann, M.; Huber, P.E.; Bischof, M.; Witt, O.; Deubzer, H.E.; Kulozik, A.E.; Debus, J.; et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Tomasovic, S.P.; Dewey, W.C. Comparative studies of the effects of drugs on X-ray-induced g2 delay. Radiat. Res. 1978, 74, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, H.; Kamada, T. A review of update clinical results of carbon ion radiotherapy. Jpn. J. Clin. Oncol. 2012, 42, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.H.; Wilder, R.B.; Wong, M.S.; Forster, K.M. Recent advances in radiotherapy for head and neck cancers. Ear Nose Throat J. 2001, 80, 704–707. [Google Scholar] [PubMed]
- Wilson, P.M.; Labonte, M.J.; Martin, S.C.; Kuwahara, S.T.; El-Khoueiry, A.; Lenz, H.J.; Ladner, R.D. Sustained inhibition of deacetylases is required for the antitumor activity of the histone deactylase inhibitors panobinostat and vorinostat in models of colorectal cancer. Investig. New Drugs 2013, 31, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hong, S.; Yoon, Y.; Hwang, S.N.; Park, J.C.; Zhang, Z.; Olson, J.J.; Hu, X.P.; Shim, H. Early prediction of response to vorinostat in an orthotopic rat glioma model. NMR Biomed. 2012, 25, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.Y.; Yap, C.W.; Wang, Z.; Ho, P.C.; Chan, S.Y.; Ng, K.Y.; Ge, Z.G.; Lin, H.S. Solubilization of vorinostat by cyclodextrins. J. Clin. Pharm. Ther. 2010, 35, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Wakimoto, N.; Yin, D.; Gery, S.; Kawamata, N.; Takai, N.; Komatsu, N.; Chumakov, A.; Imai, Y.; Koeffler, H.P. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (vorinostat, saha) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, S.; Wu, K.; Wang, B.; Wong, J.Y.; Jiang, H.; Xu, R.; Ying, L.; Huang, H.; Zheng, X.; et al. Combined effects of suberoylanilide hydroxamic acid and cisplatin on radiation sensitivity and cancer cell invasion in non-small cell lung cancer. Mol. Cancer Ther. 2016, 15, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Newhauser, W.D.; Durante, M. Assessing the risk of second malignancies after modern radiotherapy. Nat. Rev. Cancer 2011, 11, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Matsufuji, N.; Fukumura, A.; Komori, M.; Kanai, T.; Kohno, T. Influence of fragment reaction of relativistic heavy charged particles on heavy-ion radiotherapy. Phys. Med. Biol. 2003, 48, 1605–1623. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.H.; Li, J.J.; Sun, L.Q. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr. Drug Targets 2013, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, W.K.; Morton, R.A. X-ray sensitivity during the cell generation cycle of cultured chinese hamster cells. Radiat. Res. 1966, 29, 450–474. [Google Scholar] [CrossRef] [PubMed]
- Terasima, T.; Tolmach, L.J. Changes in X-ray sensitivity of hela cells during the division cycle. Nature 1961, 190, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, F.; Gnemmi, I.; Vallario, A.; Genazzani, A.A.; Canonico, P.L. Inhibitors of histone deacetylase (hdac) restore the p53 pathway in neuroblastoma cells. Br. J. Pharmacol. 2008, 153, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of γ-h2ax foci. Mol. Cancer Ther. 2006, 5, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Vallabhaneni, G.; Armstrong, E.; Huang, S.M.; Harari, P.M. Modulation of radiation response by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Chen, D.J.; Strniste, G.F. Response of X-ray-sensitive cho mutant cells to γ radiation. I. Effects of low dose rates and the process of repair of potentially lethal damage in g1 phase. Radiat. Res. 1989, 118, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Truman, A.W.; Kron, S.J.; Cote, J. Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin. Cancer Res. 2010, 16, 4543–4552. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; McAinsh, A.D.; Jackson, S.P. Saccharomyces cerevisiae sin3p facilitates DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2004, 101, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.A.; Tyler, J.K. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell Biol. 2005, 25, 4903–4913. [Google Scholar] [CrossRef] [PubMed]
- Nalabothula, N.; Carrier, F. Cancer cells’ epigenetic composition and predisposition to histone deacetylase inhibitor sensitization. Epigenomics 2011, 3, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Tobey, R.A.; Ley, K.D. Regulation of initiation of DNA synthesis in chinese hamster cells. I. Production of stable, reversible g1-arrested populations in suspension culture. J. Cell Biol. 1970, 46, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.A.; Nagasawa, H.; Weil, M.M.; Genik, P.C.; Little, J.B.; Bedford, J.S. γ-h2ax foci after low-dose-rate irradiation reveal atm haploinsufficiency in mice. Radiat. Res. 2006, 166, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Nohtomi, A.; Sakae, T.; Tsunashima, Y.; Kohno, R. Dosimetry of pulsed clinical proton beams by a small ionization chamber. Med. Phys. 2001, 28, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, J.J.; Oelfke, U. Analytical linear energy transfer calculations for proton therapy. Med. Phys. 2003, 30, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Deasy, J. Icru report 49, stopping powers and ranges for protons and alpha particles. Med. Phys. 1993, 21, 709–710. [Google Scholar] [CrossRef]
- Belli, M.; Bettega, D.; Calzolari, P.; Cherubini, R.; Cuttone, G.; Durante, M.; Esposito, G.; Furusawa, Y.; Gerardi, S.; Gialanella, G.; et al. Effectiveness of monoenergetic and spread-out bragg peak carbon-ions for inactivation of various normal and tumour human cell lines. J. Radiat. Res. 2008, 49, 597–607. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.D.; Maeda, J.; Bell, J.J.; Genet, M.D.; Phoonswadi, G.; Mann, K.A.; Kraft, S.L.; Kitamura, H.; Fujimori, A.; Yoshii, Y.; et al. Validation of 64cu-atsm damaging DNA via high-let auger electron emission. J. Radiat. Res. 2015, 56, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Roybal, E.J.; Brents, C.A.; Uesaka, M.; Aizawa, Y.; Kato, T.A. Natural and glucosyl flavonoids inhibit poly(adp-ribose) polymerase activity and induce synthetic lethality in brca mutant cells. Oncol. Rep. 2014, 31, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Bell, J.J.; Genet, S.C.; Fujii, Y.; Genet, M.D.; Brents, C.A.; Genik, P.C.; Kato, T.A. Potentially lethal damage repair in drug arrested g2-phase cells after radiation exposure. Radiat. Res. 2014, 182, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Little, J.B. Factors influencing the repair of potentially lethal radiation damage in growth-inhibited human cells. Radiat. Res. 1973, 56, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Fornace, A.J., Jr.; Nagasawa, H.; Little, J.B. Relationship of DNA repair and chromosome aberrations to potentially lethal damage repair in x-irradiated mammalian cells. Basic Life Sci. 1980, 15, 267–283. [Google Scholar] [PubMed]
- Maeda, J.; Froning, C.E.; Brents, C.A.; Rose, B.J.; Thamm, D.H.; Kato, T.A. Intrinsic radiosensitivity and cellular characterization of 27 canine cancer cell lines. PLoS ONE 2016, 11, e0156689. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irradiation Condition | A549 | AG1522 | ||||
|---|---|---|---|---|---|---|
| D10 (Gy) | SER | RBE | D10 (Gy) | SER | RBE | |
| γ-rays Control | 6.52 ± 0.33 | NA | NA | 4.42 ± 0.13 | NA | NA |
| SAHA 0.2 μM | 5.66 ± 0.44 | 1.18 ± 0.13 | NA | 4.30 ± 0.01 | 1.03 ± 0.05 | NA |
| SAHA 2 μM | 4.67 ± 0.16 | 1.43 ± 0.09 | NA | 4.08 ± 0.05 | 1.08 ± 0.03 | NA |
| Proton SOBP Control | 5.37 ± 0.24 | NA | 1.24 ± 0.07 | 3.55 ± 0.11 | NA | 1.25 ± 0.04 |
| SAHA 0.2 μM | 4.50 ± 0.01 | 1.27 ± 0.03 | 1.26 ± 0.09 | 3.16 ± 0.13 | 1.13 ± 0.05 | 1.38 ± 0.05 |
| SAHA 2 μM | 4.16 ± 0.32 | 1.31 ± 0.05 | 1.06 ± 0.09 | 3.15 ± 0.21 | 1.16 ± 0.09 | 1.33 ± 0.09 |
| Carbon ion SOBP Control | 2.56 ± 0.16 | NA | 2.59 ± 0.23 | 2.12 ± 0.04 | NA | 2.18 ± 0.31 |
| SAHA 0.2 μM | 2.24 ± 0.13 | 1.15 ± 0.16 | 2.43 ± 0.11 | 2.17 ± 0.25 | 1.05 ± 0.20 | 2.10 ± 0.32 |
| SAHA 2 μM | 2.18 ± 0.06 | 1.18 ± 0.05+ | 2.12 ± 0.04 | 2.25 ± 0.08 | 0.95 ± 0.12 | 1.82 ± 0.06 |
| AG1522, D10 (Gy) | A549, D10 (Gy) | ||||
|---|---|---|---|---|---|
| SAHA Treatment Started Subculture Time after Irradiation | 24 h Before | 24 h Before | After Irradiation | ||
| 0 h | 24 h | 0 h | 24 h | 24 h | |
| γ-rays Control | 4.31 ± 0.41 | 6.35 ± 0.44 | 6.57 ± 0.37 | 8.43 ± 0.29 | NA |
| SAHA 0.2 μM | 4.49 ± 0.45 | 6.96 ± 0.36 | 6.01 ± 0.42 | 6.97 ± 1.13 | 8.77 ± 0.41 |
| SAHA 2 μM | 4.21 ± 0.73 | 7.02 ± 0.59 | 4.85 ± 0.95 | 6.15 ± 0.59 | 8.04 ± 0.24 |
| Proton SOBP Control | 3.90 ± 0.36 | 5.35 ± 1.30 | |||
| SAHA 0.2 μM | 3.78 ± 0.47 | 5.53 ± 1.90 | |||
| SAHA 2 μM | 2.81 ± 0.39 | 4.14 ± 0.89 | |||
| Carbon ion SOBP Control | 2.11 ± 0.75 | 3.09 ± 0.13 | |||
| SAHA 0.2 μM | 2.03 ± 0.25 | 2.93 ± 0.52 | |||
| SAHA 2 μM | 1.98 ± 0.06 | 2.57 ± 0.51 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerelchuluun, A.; Maeda, J.; Manabe, E.; Brents, C.A.; Sakae, T.; Fujimori, A.; Chen, D.J.; Tsuboi, K.; Kato, T.A. Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. Int. J. Mol. Sci. 2018, 19, 496. https://doi.org/10.3390/ijms19020496
Gerelchuluun A, Maeda J, Manabe E, Brents CA, Sakae T, Fujimori A, Chen DJ, Tsuboi K, Kato TA. Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. International Journal of Molecular Sciences. 2018; 19(2):496. https://doi.org/10.3390/ijms19020496
Chicago/Turabian StyleGerelchuluun, Ariungerel, Junko Maeda, Eri Manabe, Colleen A. Brents, Takeji Sakae, Akira Fujimori, David J. Chen, Koji Tsuboi, and Takamitsu A. Kato. 2018. "Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure" International Journal of Molecular Sciences 19, no. 2: 496. https://doi.org/10.3390/ijms19020496
APA StyleGerelchuluun, A., Maeda, J., Manabe, E., Brents, C. A., Sakae, T., Fujimori, A., Chen, D. J., Tsuboi, K., & Kato, T. A. (2018). Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. International Journal of Molecular Sciences, 19(2), 496. https://doi.org/10.3390/ijms19020496