The Role of Trio, a Rho Guanine Nucleotide Exchange Factor, in Glomerular Podocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Trio mRNA Is Highly Expressed in Cultured Podocytes and Upregulated in Glomeruli in Patients with FSGS
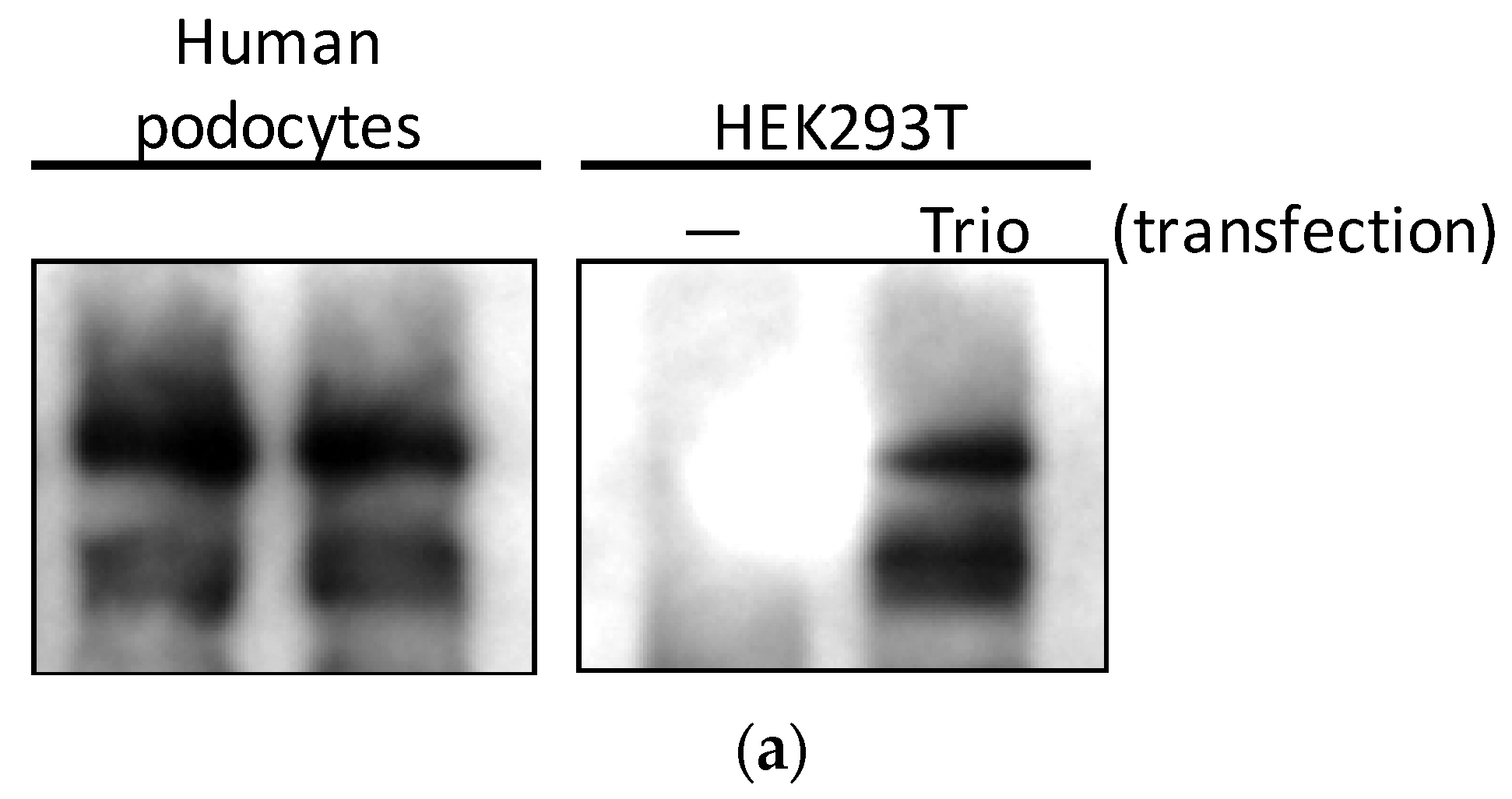
2.2. Three Isoforms of Trio Are Expressed in Podocytes
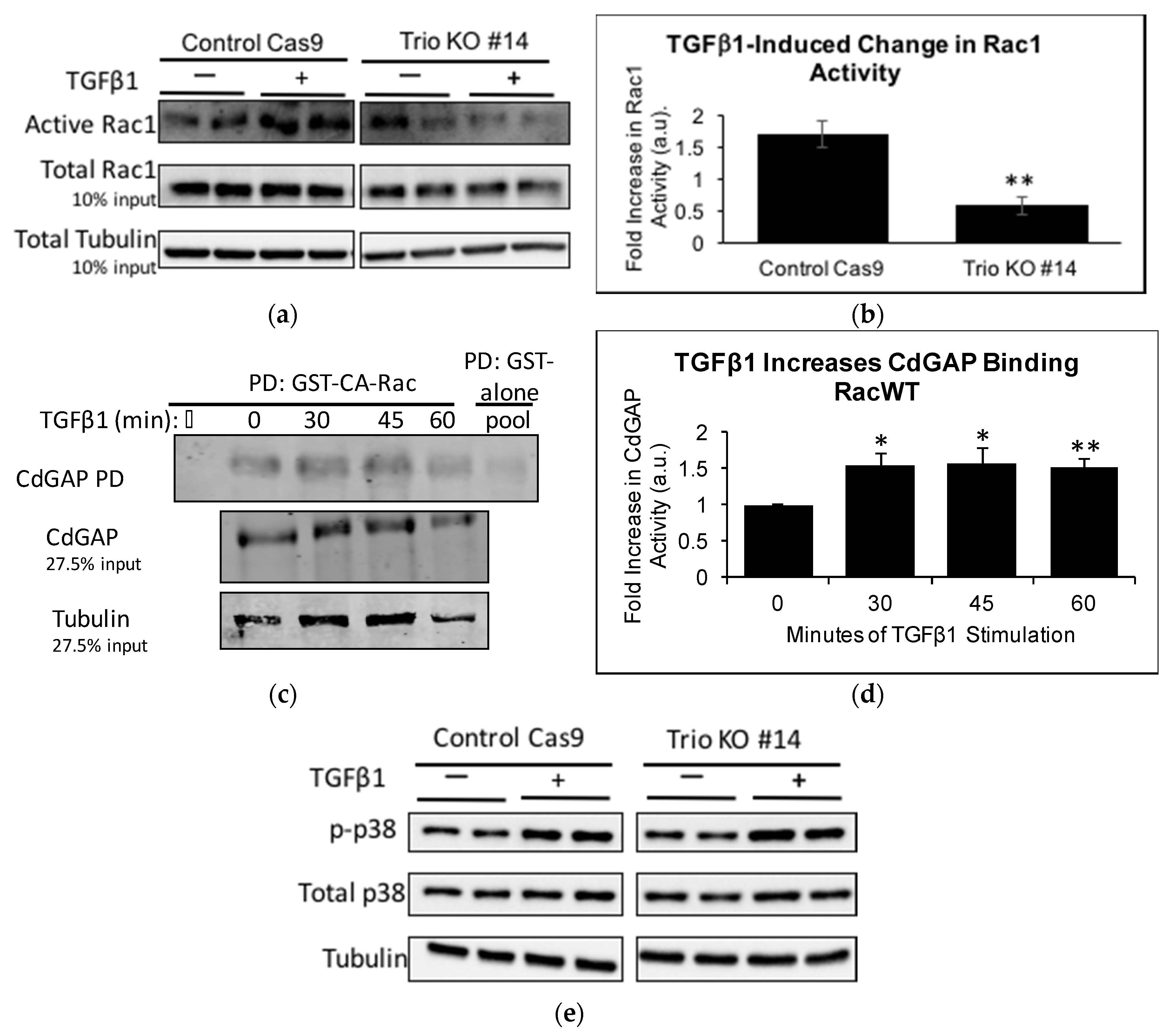
2.3. Trio Contributes to Basal Rac1 Activity and Cell Size
2.4. Trio Affects Motility, Attachment, and Vinculin Distribution of Podocytes
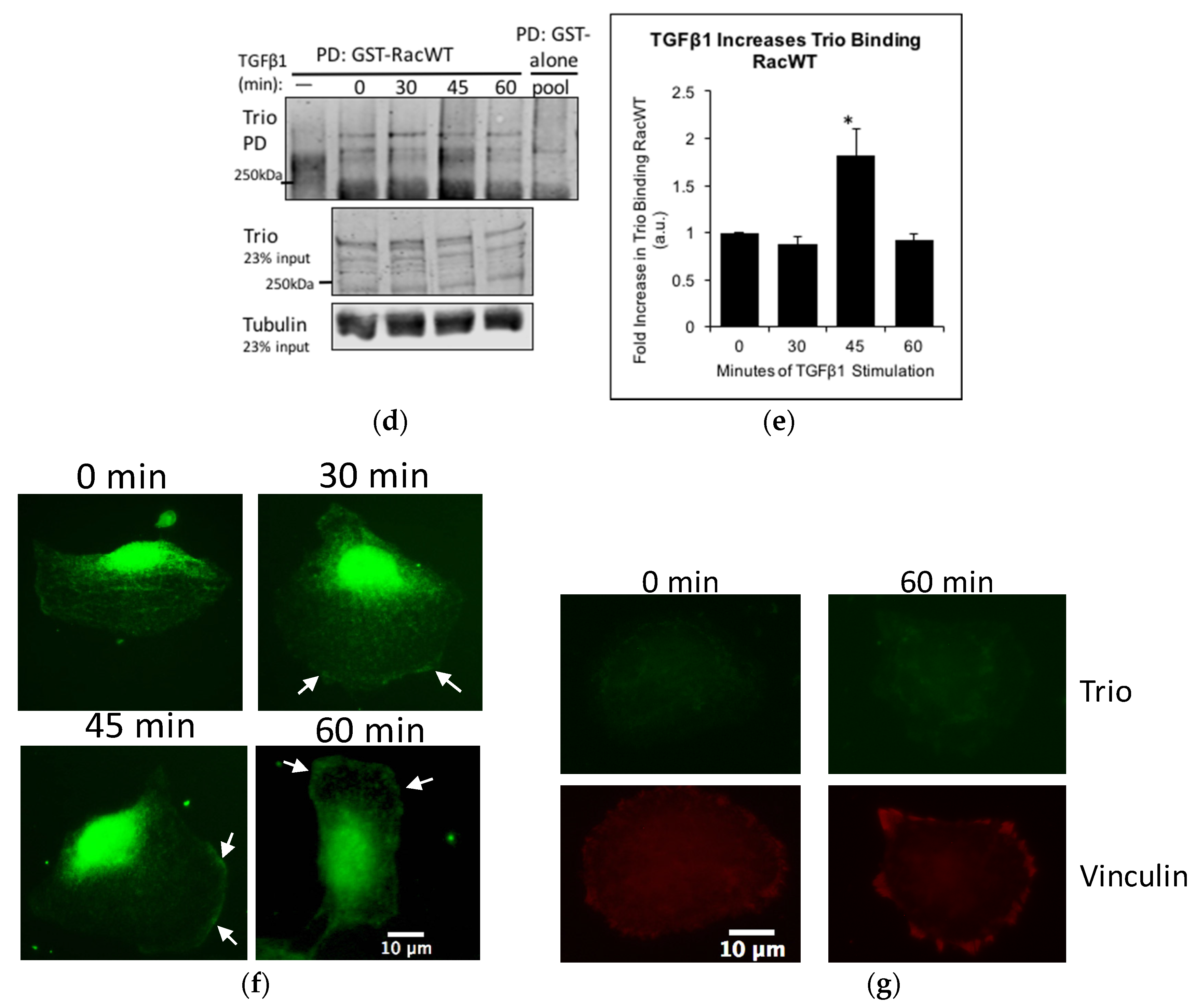
2.5. TGFβ1 Increases Trio and Rac1 Activity in Podocytes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Transfection
4.3. Generation of Trio KOs and Nucleofection
4.4. RNA-Sequencing
4.5. Nephromine/Nephroseq Analysis
4.6. Immunofluorescence Staining of Cultured Podocytes and Quantification
4.7. Immunofluorescence Staining of Paraffin-Embedded Kidney Sections
4.8. SDS-PAGE and Immunoblotting
4.9. CRIB Pulldown Assay for Active Rac1
4.10. Trio and CdGAP Pulldown
4.11. MTT Assay
4.12. Wound Healing Assay
4.13. Attachment Assay
4.14. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CA | Constitutively Active |
| CKD | Chronic Kidney Disease |
| CRIB | Cdc42/Rac Interactive Binding |
| ECL | Enhanced Chemiluminescent |
| ERCB | European renal cDNA Biobank |
| FSGS | Focal Segmental Glomerulosclerosis |
| GST | Glutathione S-transferase |
| HRP | Horseradish peroxidase |
| KO | Knockout |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen Active Protein Kinase |
| MOPS | 200 mM 3-(N-morpholino) propanesulfonic acid |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NGS | Normal Goat Serum |
| PBS | Phosphate Buffered Solution |
| PD | Pulldown |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEM | Standard Error of Mean |
| TGFβ1 | Transforming growth factor β1 |
| WT | Wild Type |
| WT1 | Wilms Tumor 1 |
References
- Maas, R.J.; Deegens, J.K.; Smeets, B.; Moeller, M.J.; Wetzels, J.F. Minimal change disease and idiopathic FSGS: Manifestations of the same disease. Nat. Rev. Nephrol. 2016, 12, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho, RAC, and CDC42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Côté, J.-F.; Gupta, I.R.; Takano, T. Rac1 activation in podocytes induces the spectrum of nephrotic syndrome. Kidney Int. 2017, 92, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Suleiman, H.; Kim, A.H.; Miner, J.H.; Dani, A.; Shaw, A.S.; Akilesh, S. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol. Cell. Biol. 2013, 33, 4755–4764. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Lee, M.-S.; Zhou, W. Dosage-dependent role of Rac1 in podocyte injury. Am. J. Physiol.-Ren. Physiol. 2016, 310, F777–F784. [Google Scholar] [CrossRef] [PubMed]
- Gupta, I.R.; Baldwin, C.; Auguste, D.; Ha, K.C.; el Andalousi, J.; Fahiminiya, S.; Bitzan, M.; Bernard, C.; Akbari, M.R.; Narod, S.A.; et al. ARHGDIA: A novel gene implicated in nephrotic syndrome. J. Med. Genet. 2013, 50, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Saisawat, P.; Ashraf, S.; Hurd, T.W.; Vega-Warner, V.; Fang, H.; Beck, B.B.; Gribouval, O.; Zhou, W.; Diaz, K.A.; et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Investig. 2013, 123, 3243–3253. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Takano, T.; Sapir-Pichhadze, R. Changing paradigms in the management of rejection in kidney transplantation: Evolving from protocol-based care to the Era of P4 medicine. Can. J. Kidney Health Dis. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Suleiman, H.; Yu, H.; Stander, M.C.; Lavin, P.; Gbadegesin, R.; Antignac, C.; Pollak, M.; Kopp, J.B.; Winn, M.P.; et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Investig. 2011, 121, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.-C.; He, J.C.; Wang, Z.-H.; Feng, X.; Fukumi-Tominaga, T.; Chen, N.; Xu, J.; Iyengar, R.; Klotman, P.E. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J. Biol. Chem. 2008, 283, 8173–8182. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-Y.; Ng, K.-H.; Chen, J.; Lu, J.; Lee, C.G.; Tan, P.-H.; Jordan, S.C.; He, H.Y.; Yap, H.K. Novel role of Vav1-Rac1 pathway in actin cytoskeleton regulation in interleukin-13-induced minimal change-like nephropathy. Clin. Sci. 2016, 130, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Laurin, M.; Dumouchel, A.; Fukui, Y.; Côté, J.-F. The Rac-specific exchange factors Dock1 and Dock5 are dispensable for the establishment of the glomerular filtration barrier in vivo. Small GTPases 2013, 4, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Van Rijssel, J.; van Buul, J.D. The many faces of the guanine-nucleotide exchange factor trio. Cell Adhes. Migr. 2012, 6, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Van Rijssel, J.; Hoogenboezem, M.; Wester, L.; Hordijk, P.L.; van Buul, J.D. The N-terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS ONE 2012, 7, e29912. [Google Scholar] [CrossRef] [PubMed]
- Debant, A.; Serra-Pagès, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.-H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e983880. [Google Scholar] [CrossRef] [PubMed]
- Attias, O.; Jiang, R.; Aoudjit, L.; Kawachi, H.; Takano, T. Rac1 contributes to actin organization in glomerular podocytes. Nephron Exp. Nephrol. 2009, 114, e93–e106. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Tran, N.L.; Chan, A.; Wolf, A.; Nakada, M.; Rutka, F.; Ennis, M.; McDonough, W.S.; Berens, M.E.; Symons, M.; et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am. J. Pathol. 2008, 173, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Garcia-Mata, R.; Burridge, K. Rho protein crosstalk: Another social network? Trends Cell Biol. 2011, 21, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zou, L.-X.; Chen, M.-J. Make precision medicine work for chronic kidney disease. Med. Princ. Pract. 2017, 26, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, B.K.; Moon, K.C.; Hong, H.K.; Lee, H.S. Activation of the TGF-β/SMAD signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 2003, 64, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Hubchak, S.C.; Sparks, E.E.; Hayashida, T.; Schnaper, H.W. Rac1 promotes TGF-β-stimulated mesangial cell type I collagen expression through a PI3K/Akt-dependent mechanism. Am. J. Physiol.-Ren. Physiol. 2009, 297, F1316–F1323. [Google Scholar] [CrossRef] [PubMed]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Gupta, I.R.; Takano, T. Loss of Rho-GDIα sensitizes podocytes to lipopolysaccharide-mediated injury. Am. J. Physiol. Ren. Physiol. 2015, 308, F1207–F1216. [Google Scholar] [CrossRef] [PubMed]
- García-Mata, R.; Wennerberg, K.; Arthur, W.T.; Noren, N.K.; Ellerbroek, S.M.; Burridge, K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006, 406, 425–437. [Google Scholar] [PubMed]
- He, Y.; Northey, J.; Primeau, M.; Machado, R.; Trembath, R.; Siegel, P.; Lamarche-Vane, N. CdGAP is required for transforming growth factor β-and Neu/ErbB-2-induced breast cancer cell motility and invasion. Oncogene 2011, 30, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Auguste, D.; Maier, M.; Baldwin, C.; Aoudjit, L.; Robins, R.; Gupta, I.R.; Takano, T. Disease-causing mutations of RhoGDIα induce Rac1 hyperactivation in podocytes. Small GTPases 2016, 7, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Song, C.Y. Effects of TGF-β on podocyte growth and disease progression in proliferative podocytopathies. Kidney Blood Press. Res. 2010, 33, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Factor, V.M.; Mozes, M.; Nagy, P.; Sanderson, N.; Böttinger, E.P.; Klotman, P.E.; Thorgeirsson, S.S. Transgenic mice with increased plasma levels of TGF-β1 develop progressive renal disease. Lab. Investig. J. Tech. Methods Pathol. 1996, 74, 991–1003. [Google Scholar]
- Schiffer, M.; Bitzer, M.; Roberts, I.S.; Kopp, J.B.; ten Dijke, P.; Mundel, P.; Böttinger, E.P. Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Investig. 2001, 108, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Niranjan, T.; Krick, S.; Zhang, T.; Ju, W.; Shaw, A.S.; Schiffer, M.; Böttinger, E.P. TβRI independently activates Smad-and CD2AP-dependent pathways in podocytes. J. Am. Soc. Nephrol. 2009, 20, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Wahab, N.; Schaefer, L.; Weston, B.; Yiannikouris, O.; Wright, A.; Babelova, A.; Schaefer, R.; Mason, R.M. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia 2005, 48, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Noble, N.A.; Cohen, A.H.; Nast, C.C.; Hishida, A.; Gold, L.I.; Border, W.A. Expression of transforming growth factor-β isoforms in human glomerular diseases. Kidney Int. 1996, 49, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Clarkson, M.R.; Duggan, J.; Brady, H.R. Connective tissue growth factor: Potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000, 58, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Seipel, K.; Medley, Q.G.; Kedersha, N.L.; Zhang, X.A.; O’Brien, S.P.; Serra-Pages, C.; Hemler, M.E.; Streuli, M. Trio amino-terminal guanine nucleotide exchange factor domain expression promotes actin cytoskeleton reorganization, cell migration and anchorage-independent cell growth. J. Cell Sci. 1999, 112, 1825–1834. [Google Scholar] [PubMed]
- Chen, S.-Y.; Huang, P.-H.; Cheng, H.-J. Disrupted-in-Schizophrenia 1–mediated axon guidance involves TRIO-RAC-PAK small GTPase pathway signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 5861–5866. [Google Scholar] [CrossRef] [PubMed]
- Neubrand, V.E.; Thomas, C.; Schmidt, S.; Debant, A.; Schiavo, G. Kidins220/ARMS regulates Rac1-dependent neurite outgrowth by direct interaction with the RhoGEF Trio. J. Cell Sci. 2010, 123, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Seipel, K.; O’Brien, S.P.; Iannotti, E.; Medley, Q.G.; Streuli, M. Tara, a novel F-actin binding protein, associates with the Trio guanine nucleotide exchange factor and regulates actin cytoskeletal organization. J. Cell Sci. 2001, 114, 389–399. [Google Scholar] [PubMed]
- Yano, T.; Yamazaki, Y.; Adachi, M.; Okawa, K.; Fort, P.; Uji, M.; Tsukita, S.; Tsukita, S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J. Cell Biol. 2011, 193, 319–332. [Google Scholar] [CrossRef] [PubMed]
- DeGeer, J.; Kaplan, A.; Mattar, P.; Morabito, M.; Stochaj, U.; Kennedy, T.E.; Debant, A.; Cayouette, M.; Fournier, A.E.; Lamarche-Vane, N. Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J. Cell Biol. 2015, 210, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, I.M.; Hightower, L.E. Transforming growth factor-β1 rapidly induces Hsp70 and Hsp90 molecular chaperones in cultured chicken embryo cells. J. Cell. Physiol. 1992, 152, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Maier, M. (McGill University, Montreal, QC, USA). Human Podocytes Treated with TGFβ1 and Immunoblotted for Hsc70. Personal conmmunication, 2017. [Google Scholar]
- Zhang, H.; Cybulsky, A.V.; Aoudjit, L.; Zhu, J.; Li, H.; Lamarche-Vane, N.; Takano, T. Role of Rho-GTPases in complement-mediated glomerular epithelial cell injury. Am. J. Physiol.-Ren. Physiol. 2007, 293, F148–F156. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [PubMed]
- Mundel, P.; Reiser, J.; Borja, A.Z.M.; Pavenstädt, H.; Davidson, G.R.; Kriz, W.; Zeller, R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 1997, 236, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Kachurina, N.; Chung, C.-F.; Benderoff, E.; Babayeva, S.; Bitzan, M.; Goodyer, P.; Kitzler, T.; Matar, D.; Cybulsky, A.V.; Alachkar, N.; et al. Novel unbiased assay for circulating podocyte-toxic factors associated with recurrent focal segmental glomerulosclerosis. Am. J. Physiol.-Ren. Physiol. 2016, 310, F1148–F1156. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maier, M.; Baldwin, C.; Aoudjit, L.; Takano, T. The Role of Trio, a Rho Guanine Nucleotide Exchange Factor, in Glomerular Podocytes. Int. J. Mol. Sci. 2018, 19, 479. https://doi.org/10.3390/ijms19020479
Maier M, Baldwin C, Aoudjit L, Takano T. The Role of Trio, a Rho Guanine Nucleotide Exchange Factor, in Glomerular Podocytes. International Journal of Molecular Sciences. 2018; 19(2):479. https://doi.org/10.3390/ijms19020479
Chicago/Turabian StyleMaier, Mirela, Cindy Baldwin, Lamine Aoudjit, and Tomoko Takano. 2018. "The Role of Trio, a Rho Guanine Nucleotide Exchange Factor, in Glomerular Podocytes" International Journal of Molecular Sciences 19, no. 2: 479. https://doi.org/10.3390/ijms19020479
APA StyleMaier, M., Baldwin, C., Aoudjit, L., & Takano, T. (2018). The Role of Trio, a Rho Guanine Nucleotide Exchange Factor, in Glomerular Podocytes. International Journal of Molecular Sciences, 19(2), 479. https://doi.org/10.3390/ijms19020479