Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity


Abstract
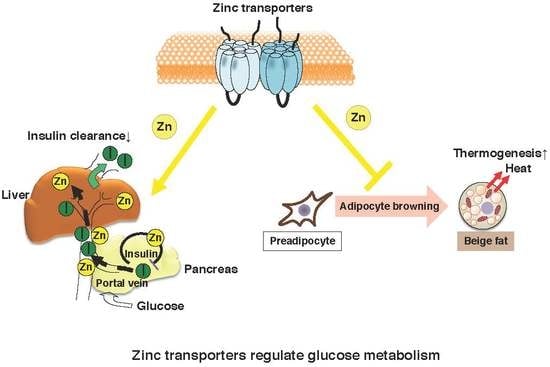
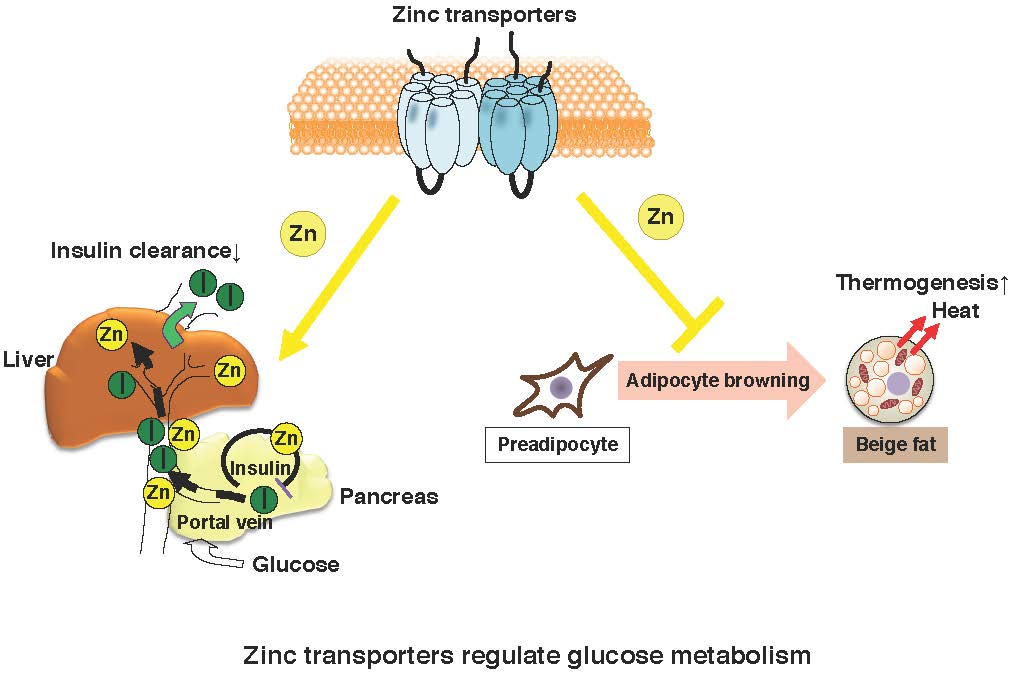{kind=link}
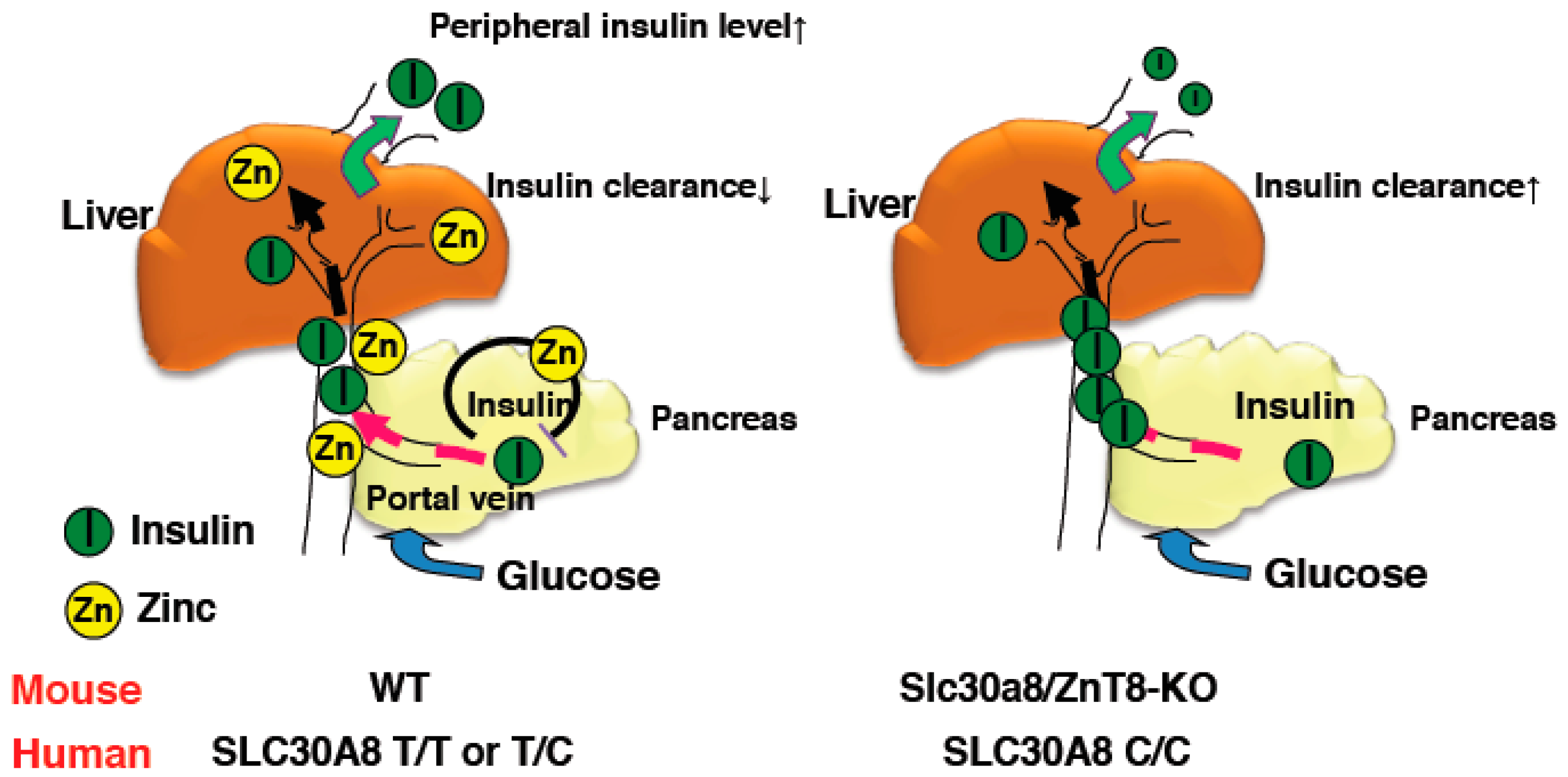{kind=link}
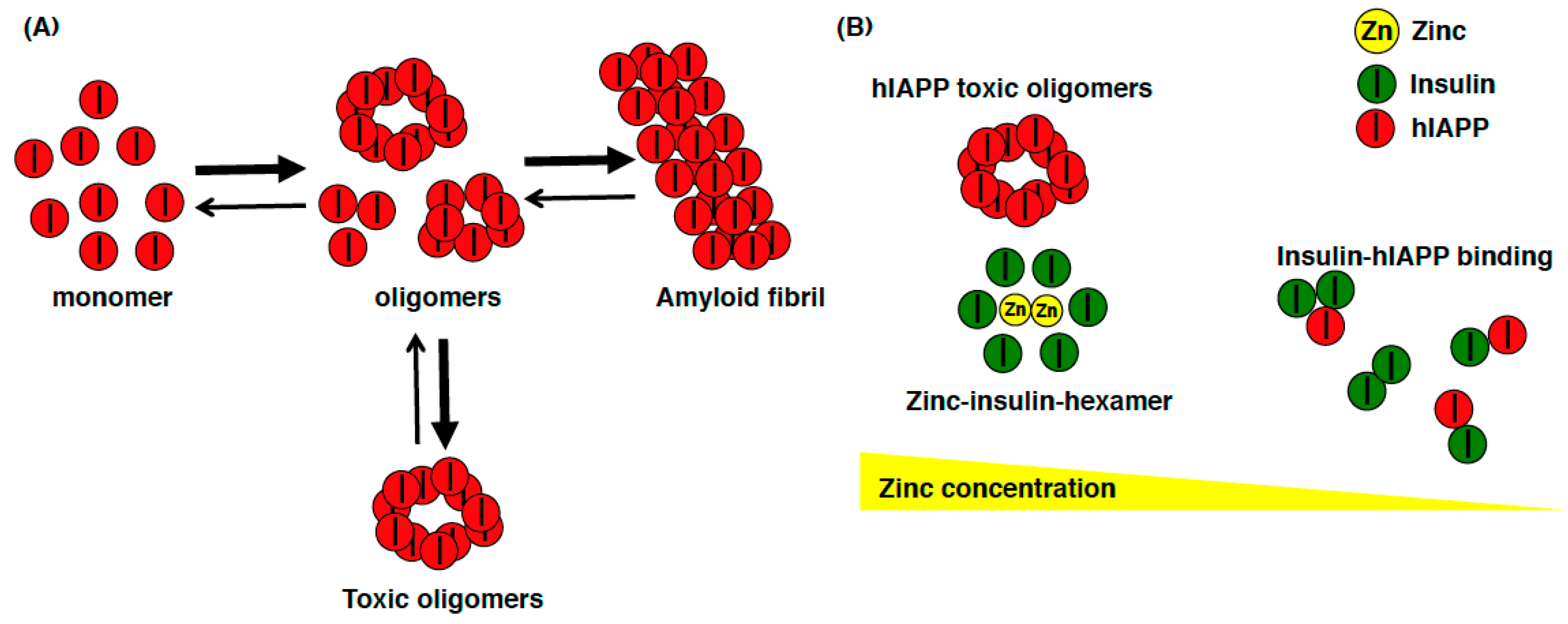{kind=link}
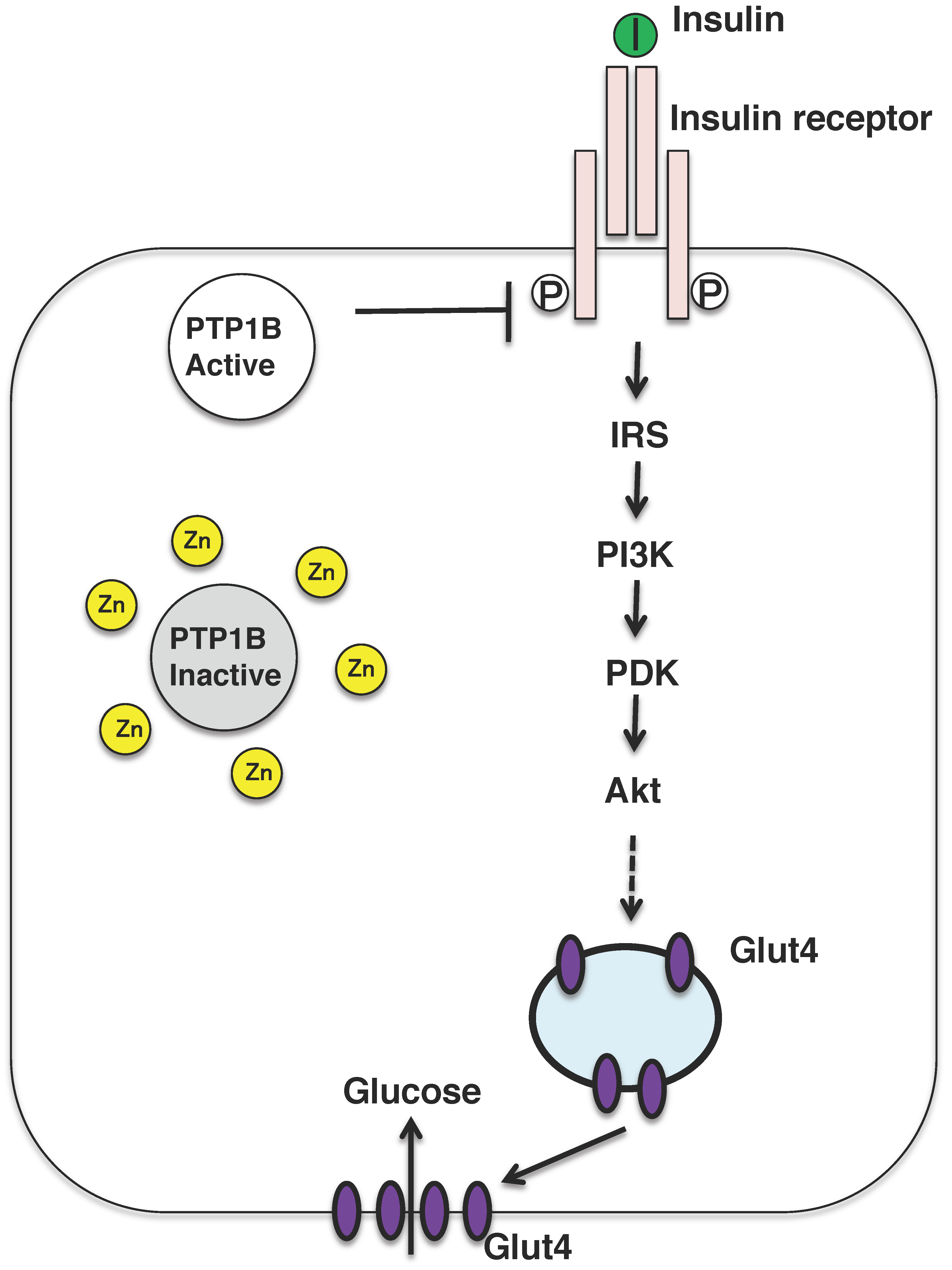{kind=link}
{kind=link}
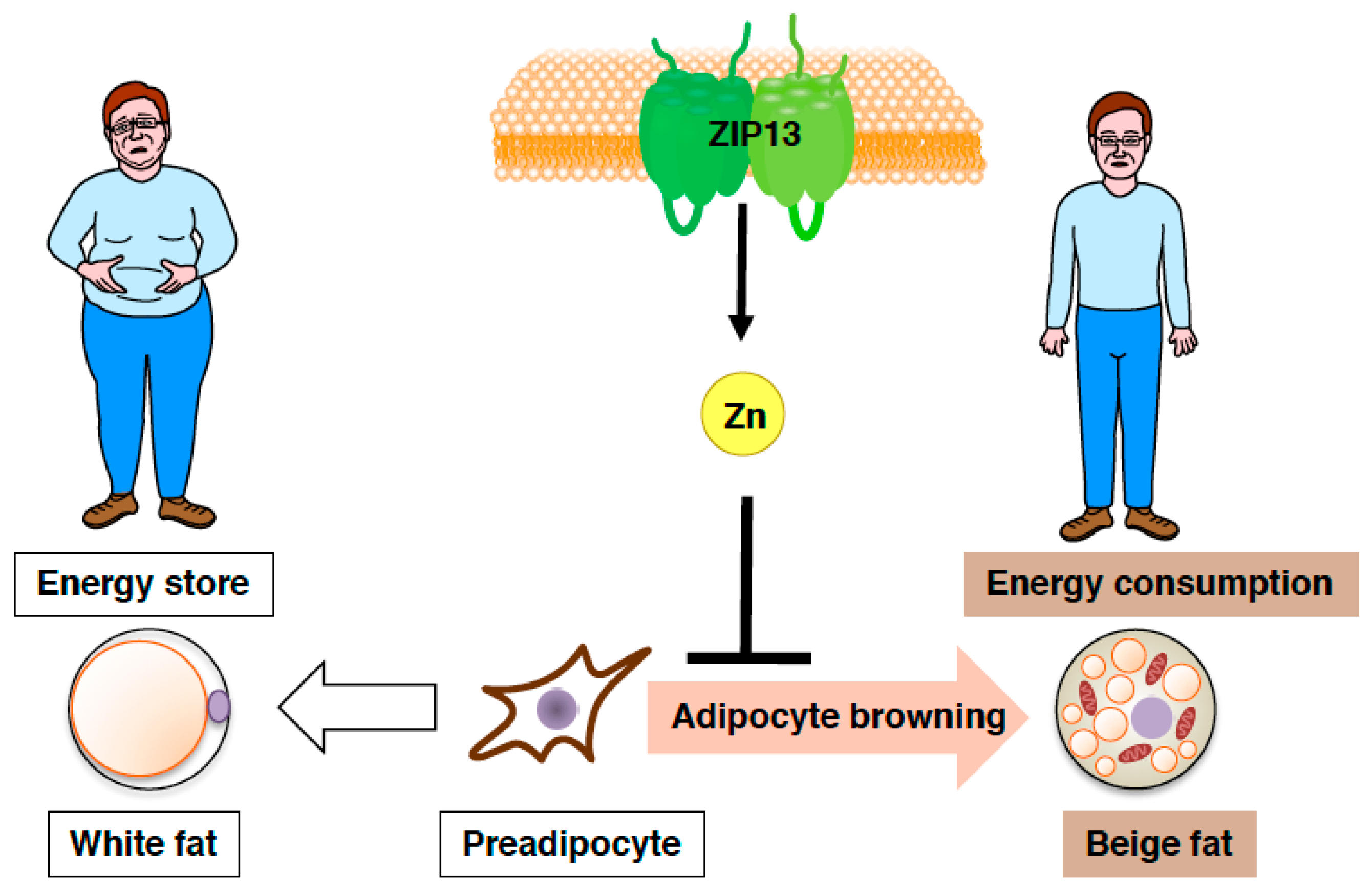{kind=link}
1. Introduction
2. Zinc Homeostasis and Pancreatic β Cells
2.1. Insulin Biosynthesis in Pancreatic β Cells
2.2. Zinc Supplementation in Diabetic Animals and Patients
3. ZnT8 Plays a Crucial Role in Glucose Homeostasis
3.1. Insulin Secretory Granule of ZnT8-KO Mice
3.2. Phenotypes of ZnT8-KO Mice Regarding Glucose Metabolism
3.3. Involvement of Other ZnT Transporters
3.4. Zinc Transport Activity of ZnT8 Variants
4. Zinc Distribution Affects Adipocyte Metabolism
4.1. Zinc Distribution in Obesity
4.2. Association between Adipocyte Metabolism and Zinc Homeostasis
4.3. ZIP13 Regulates Beige Adipocyte Biogenesis and Energy Expenditure
5. Involvement of Zinc in the Insulin Signaling Pathway
5.1. Zinc Transporters Affect Skeletal Muscle Insulin Signaling
5.2. Zinc Homeostasis and Sarcopenia
6. Summary
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Simcox, J.A.; McClain, D.A. Iron and diabetes risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in Pancreatic Islet Biology, Insulin Sensitivity, and Diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Leapman, R.D.; Li, M.X.; Atwater, I. Elemental composition of secretory granules in pancreatic islets of Langerhans. Biophys. J. 1993, 64, 525–532. [Google Scholar] [CrossRef]
- Hutton, J.C.; Penn, E.J.; Peshavaria, M. Low-molecular-weight constituents of isolated insulin-secretory granules. Bivalent cations, adenine nucleotides and inorganic phosphate. Biochem. J. 1983, 210, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takeda, T.A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of zinc transporters: Molecular and genetic importance in zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, F.; Devergnas, S.; Pattou, F.; Schuit, F.; Garcia-Cuenca, R.; Vandewalle, B.; Kerr-Conte, J.; Van Lommel, L.; Grunwald, D.; Favier, A.; et al. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J. Cell Sci. 2006, 119, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.; Steiner, D. The role of assembly in insulin’s biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194. [Google Scholar] [CrossRef]
- Dodson, E.J.; Dodson, G.G.; Hodgkin, D.C.; Reynolds, C.D. Structural relationships in the two-zinc insulin hexamer. Can. J. Biochem. 1979, 57, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.F. Zinc-ligand interactions modulate assembly and stability of the insulin hexamer—A review. Biometals 2005, 18, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Havu, N.; Lundgren, G.; Falkmer, S. Zinc and manganese contents of micro-dissected pancreatic islets of some rodents. A microchemical study in adult and newborn guinea pigs, rats, Chinese hamsters and spiny mice. Acta Endocrinol. 1977, 86, 570–577. [Google Scholar] [PubMed]
- Steiner, D.F.; Rouille, Y.; Gong, Q.; Martin, S.; Carroll, R.; Chan, S.J. The role of prohormone convertases in insulin biosynthesis: Evidence for inherited defects in their action in man and experimental animals. Diabetes Metab. 1996, 22, 94–104. [Google Scholar] [PubMed]
- Emdin, S.O.; Dodson, G.G.; Cutfield, J.M.; Cutfield, S.M. Role of zinc in insulin biosynthesis. Some possible zinc-insulin interactions in the pancreatic B-cell. Diabetologia 1980, 19, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hutton, J.C. The insulin secretory granule. Diabetologia 1989, 32, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.C.; Min, L.; Pessin, J.E. Insulin granule biogenesis, trafficking and exocytosis. Vitam. Horm. 2009, 80, 473–506. [Google Scholar] [PubMed]
- Tamaki, M.; Fujitani, Y.; Hara, A.; Uchida, T.; Tamura, Y.; Takeno, K.; Kawaguchi, M.; Watanabe, T.; Ogihara, T.; Fukunaka, A.; et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J. Clin. Investig. 2013, 123, 4513–4524. [Google Scholar] [CrossRef] [PubMed]
- Boquist, L.; Lernmark, A. Effects on the endocrine pancreas in Chinese hamsters fed zinc deficient diets. Acta Pathol. Microbiol. Scand. 1969, 76, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.M.; Gershoff, S.N. Effect of zinc deficiency in rats on insulin release from the pancreas. J. Nutr. 1973, 103, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005, 54 (Suppl. S2), S108–S113. [Google Scholar] [CrossRef] [PubMed]
- Mysore, T.B.; Shinkel, T.A.; Collins, J.; Salvaris, E.J.; Fisicaro, N.; Murray-Segal, L.J.; Johnson, L.E.; Lepore, D.A.; Walters, S.N.; Stokes, R.; et al. Overexpression of glutathione peroxidase with two isoforms of superoxide dismutase protects mouse islets from oxidative injury and improves islet graft function. Diabetes 2005, 54, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.L.; Westman, N.G.; Lundgren, E.; Roos, G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982, 42, 1955–1961. [Google Scholar] [PubMed]
- Sun, Q.; van Dam, R.M.; Willett, W.C.; Hu, F.B. Prospective study of zinc intake and risk of type 2 diabetes in women. Diabetes Care 2009, 32, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Bao, W.; Zhang, Y.; Rong, Y.; Wang, X.; Jin, Y.; Song, Y.; Yao, P.; Sun, C.; Hu, F.B.; et al. Interactions between zinc transporter-8 gene (SLC30A8) and plasma zinc concentrations for impaired glucose regulation and type 2 diabetes. Diabetes 2014, 63, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Sun, W.; Fu, Y.; Miao, L.; Cai, L. Zinc homeostasis in the metabolic syndrome and diabetes. Front. Med. 2013, 7, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular zinc in insulin secretion and action: A determinant of diabetes risk? Proc. Nutr. Soc. 2016, 75, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Chabosseau, P.; Rutter, G.A. Zinc and diabetes. Arch. Biochem. Biophys. 2016, 611, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Dai, F.F.; Hardy, A.B.; Giglou, P.R.; Bhattacharjee, A.; Koshkin, V.; Chimienti, F.; Gaisano, H.Y.; Rutter, G.A.; Wheeler, M.B. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010, 53, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.K.; Hu, M.; Chabosseau, P.L.; Cane, M.C.; Meur, G.; Bellomo, E.A.; Carzaniga, R.; Collinson, L.M.; Li, W.H.; Hodson, D.J.; et al. Molecular Genetic Regulation of SLC30A8/ZnT8 Reveals a Positive Association With Glucose Tolerance. Mol. Endocrinol. 2016, 30, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Pound, L.D.; Sarkar, S.A.; Ustione, A.; Dadi, P.K.; Shadoan, M.K.; Lee, C.E.; Walters, J.A.; Shiota, M.; McGuinness, O.P.; Jacobson, D.A.; et al. The physiological effects of deleting the mouse SLC30A8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS ONE 2012, 7, e40972. [Google Scholar] [CrossRef] [PubMed]
- Pound, L.D.; Sarkar, S.A.; Benninger, R.K.; Wang, Y.; Suwanichkul, A.; Shadoan, M.K.; Printz, R.L.; Oeser, J.K.; Lee, C.E.; Piston, D.W.; et al. Deletion of the mouse SLC30A8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem. J. 2009, 421, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Holst, J.J.; Schmidt, W.E.; Nauck, M.A. Reduction of hepatic insulin clearance after oral glucose ingestion is not mediated by glucagon-like peptide 1 or gastric inhibitory polypeptide in humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E849–E856. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Yang, Y.; Rezaei, K.; Fernstrom, M.A.; Lee, A.D.; Kido, Y.; Erickson, S.K.; Najjar, S.M. CEACAM1 regulates insulin clearance in liver. Nat. Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Boesgaard, T.W.; Zilinskaite, J.; Vanttinen, M.; Laakso, M.; Jansson, P.A.; Hammarstedt, A.; Smith, U.; Stefan, N.; Fritsche, A.; Haring, H.; et al. The common SLC30A8 Arg325Trp variant is associated with reduced first-phase insulin release in 846 non-diabetic offspring of type 2 diabetes patients—The EUGENE2 study. Diabetologia 2008, 51, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Malito, E.; Hedouin, S.; Reinstatler, L.; Sahara, T.; Abdul-Hay, S.O.; Choudhry, S.; Maharvi, G.M.; Fauq, A.H.; Huzarska, M.; et al. Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS ONE 2010, 5, e10504. [Google Scholar] [CrossRef] [PubMed]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, H.J.; Cole, T.B.; Born, D.E.; Schwartzkroin, P.A.; Palmiter, R.D. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc. Natl. Acad. Sci. USA 1997, 94, 12676–12681. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose regulates free cytosolic Zn2+ concentration, Slc39 (ZiP), and metallothionein gene expression in primary pancreatic islet beta-cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef] [PubMed]
- Smidt, K.; Jessen, N.; Petersen, A.B.; Larsen, A.; Magnusson, N.; Jeppesen, J.B.; Stoltenberg, M.; Culvenor, J.G.; Tsatsanis, A.; Brock, B.; et al. SLC30A3 responds to glucose- and zinc variations in beta-cells and is critical for insulin production and in vivo glucose-metabolism during beta-cell stress. PLoS ONE 2009, 4, e5684. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Narita, H.; Yamaguchi-Iwai, Y.; Hirose, J.; Amano, T.; Sugiura, N.; Sasaki, R.; Mori, K.; Iwanaga, T.; Nagao, M. Cloning and characterization of a novel mammalian zinc transporter, zinc transporter 5, abundantly expressed in pancreatic beta cells. J. Biol. Chem. 2002, 277, 19049–19055. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yan, M.; Kirschke, C.P. Over-expression of ZnT7 increases insulin synthesis and secretion in pancreatic beta-cells by promoting insulin gene transcription. Exp. Cell Res. 2010, 316, 2630–2643. [Google Scholar] [CrossRef] [PubMed]
- Syring, K.E.; Boortz, K.A.; Oeser, J.K.; Ustione, A.; Platt, K.A.; Shadoan, M.K.; McGuinness, O.P.; Piston, D.W.; Powell, D.R.; O’Brien, R.M. Combined Deletion of Slc30a7 and SLC30A8 Unmasks a Critical Role for ZnT8 in Glucose-Stimulated Insulin Secretion. Endocrinology 2016, 157, 4534–4541. [Google Scholar] [CrossRef] [PubMed]
- Merriman, C.; Huang, Q.; Rutter, G.A.; Fu, D. Lipid-tuned Zinc Transport Activity of Human ZnT8 Protein Correlates with Risk for Type-2 Diabetes. J. Biol. Chem. 2016, 291, 26950–26957. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.P.; Allen, N.B.; Meyers, M.S.; Link, E.O.; Zhang, X.; MacRenaris, K.W.; El Muayed, M. Exploring the Association Between Demographics, SLC30A8 Genotype, and Human Islet Content of Zinc, Cadmium, Copper, Iron, Manganese and Nickel. Sci. Rep. 2017, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chimienti, F. SLC30A8 mutations in type 2 diabetes. Diabetologia 2015, 58, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Langen, R.; Gurlo, T.; Matveyenko, A.V.; Butler, P.C. beta-Cell failure in type 2 diabetes: A case of asking too much of too few? Diabetes 2013, 62, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Nedumpully-Govindan, P.; Ding, F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci. Rep. 2015, 5, 8240. [Google Scholar] [CrossRef] [PubMed]
- Hashemipour, M.; Kelishadi, R.; Shapouri, J.; Sarrafzadegan, N.; Amini, M.; Tavakoli, N.; Movahedian-Attar, A.; Mirmoghtadaee, P.; Poursafa, P. Effect of zinc supplementation on insulin resistance and components of the metabolic syndrome in prepubertal obese children. Hormones 2009, 8, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Protein tyrosine phosphatases as targets of the combined insulinomimetic effects of zinc and oxidants. Biometals 2005, 18, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in Cellular Regulation: The Nature and Significance of “Zinc Signals”. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Kahn, C.R. Brown fat as a therapy for obesity and diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Troche, C.; Aydemir, T.B.; Cousins, R.J. Zinc transporter Slc39a14 regulates inflammatory signaling associated with hypertrophic adiposity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E258–E268. [Google Scholar] [CrossRef] [PubMed]
- Maxel, T.; Smidt, K.; Larsen, A.; Bennetzen, M.; Cullberg, K.; Fjeldborg, K.; Lund, S.; Pedersen, S.B.; Rungby, J. Gene expression of the zinc transporter ZIP14 (SLC39a14) is affected by weight loss and metabolic status and associates with PPARgamma in human adipose tissue and 3T3-L1 pre-adipocytes. BMC Obes. 2015, 2, 46. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Saito, M. A new era in brown adipose tissue biology: Molecular control of brown fat development and energy homeostasis. Annu. Rev. Physiol. 2014, 76, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, X.; Tang, Q.Q. Transcriptional regulation of adipocyte differentiation: A central role for CCAAT/enhancer-binding protein (C/EBP) beta. J. Biol. Chem. 2015, 290, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Kagata, T.; Johmura, Y.; Hishida, T.; Nishizuka, M.; Imagawa, M. SLC39A14, a LZT protein, is induced in adipogenesis and transports zinc. FEBS J. 2005, 272, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Beyersmann, D. Transient peaks in zinc and metallothionein levels during differentiation of 3T3L1 cells. Arch. Biochem. Biophys. 1999, 364, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tepaamorndech, S.; Kirschke, C.P.; Pedersen, T.L.; Keyes, W.R.; Newman, J.W.; Huang, L. Zinc transporter 7 deficiency affects lipid synthesis in adipocytes by inhibiting insulin-dependent Akt activation and glucose uptake. FEBS J. 2016, 283, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Seale, P. Transcriptional Regulatory Circuits Controlling Brown Fat Development and Activation. Diabetes 2015, 64, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Fukunaka, A.; Fukada, T.; Bhin, J.; Suzuki, L.; Tsuzuki, T.; Takamine, Y.; Bin, B.H.; Yoshihara, T.; Ichinoseki-Sekine, N.; Naito, H.; et al. Zinc transporter ZIP13 suppresses beige adipocyte biogenesis and energy expenditure by regulating C/EBP-beta expression. PLoS Genet. 2017, 13, e1006950. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, K.; Luijten, I.H.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Physiology of Zinc: General aspects. In Zinc in Human Biology; Mills, C.F., Ed.; Springer: London, UK, 1989; pp. 1–14. [Google Scholar]
- Myers, S.A.; Nield, A.; Chew, G.S.; Myers, M.A. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PLoS ONE 2013, 8, e79316. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kirschke, C.P.; Lay, Y.A.; Levy, L.B.; Lamirande, D.E.; Zhang, P.H. Znt7-null mice are more susceptible to diet-induced glucose intolerance and insulin resistance. J. Biol. Chem. 2012, 287, 33883–33896. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Fukunaka, A.; Hagihara, M.; Watanabe, K.; Kamino, S.; Kambe, T.; Enomoto, S.; Hiromura, M. Essential role of the zinc transporter ZIP9/SLC39A9 in regulating the activations of Akt and Erk in B-cell receptor signaling pathway in DT40 cells. PLoS ONE 2013, 8, e58022. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci. Signal. 2012, 5, ra11. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef] [PubMed]
- Cleasby, M.E.; Jamieson, P.M.; Atherton, P.J. Insulin resistance and sarcopenia: Mechanistic links between common co-morbidities. J. Endocrinol. 2016, 229, R67–R81. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Bouzan, A.; Pierrel, E.; Melly, S.; Stauffer, D.; Gutzwiller, S.; Nolin, E.; Dornelas, C.; Fryer, C.; Leighton-Davies, J.; et al. Blockade of Metallothioneins 1 and 2 Increases Skeletal Muscle Mass and Strength. Mol. Cell. Biol. 2017, 37, e00305–e00316. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Troche, C.; Kim, J.; Kim, M.H.; Teran, O.Y.; Leeuwenburgh, C.; Cousins, R.J. Aging amplifies multiple phenotypic defects in mice with zinc transporter Zip14 (Slc39a14) deletion. Exp. Gerontol. 2016, 85, 88–94. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukunaka, A.; Fujitani, Y. Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. Int. J. Mol. Sci. 2018, 19, 476. https://doi.org/10.3390/ijms19020476
Fukunaka A, Fujitani Y. Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. International Journal of Molecular Sciences. 2018; 19(2):476. https://doi.org/10.3390/ijms19020476
Chicago/Turabian StyleFukunaka, Ayako, and Yoshio Fujitani. 2018. "Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity" International Journal of Molecular Sciences 19, no. 2: 476. https://doi.org/10.3390/ijms19020476
APA StyleFukunaka, A., & Fujitani, Y. (2018). Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. International Journal of Molecular Sciences, 19(2), 476. https://doi.org/10.3390/ijms19020476