Abstract
Following injury, corneal stromal keratocytes transform into repair-phenotype of activated stromal fibroblasts (SFs) and participate in wound repair. Simultaneously, ongoing bi-directional communications between corneal stromal-epithelial cells also play a vital role in mediating the process of wound healing. Factors produced by stromal cells are known to induce proliferation, differentiation, and motility of corneal epithelial cells, which are also subsequently the main processes that occur during wound healing. In this context, the present study aims to investigate the effect of SFs conditioned medium (SFCM) on corneal epithelial cell function along with substance P (SP). Antibody microarrays were employed to profile differentially expressed cell surface markers and cytokines in the presence of SFCM and SP. Antibody microarray data revealed enhanced expression of the ITGB1 in corneal epithelial cells following stimulation with SP whereas SFCM induced abundant expression of IL-8, ITGB1, PD1L1, PECA1, IL-15, BDNF, ICAM1, CD8A, CD44 and NTF4. All these proteins have either direct or indirect roles in epithelial cell growth, movement and adhesion related signaling cascades during tissue regeneration. We also observed activation of MAPK signaling pathway along with increased expression of focal adhesion kinase (FAK), paxillin, vimentin, β-catenin and vasodilator-stimulated phosphoprotein (VASP) phosphorylation. Additionally, epithelial-to-mesenchymal transition (EMT) regulating transcription factors Slug and ZEB1 expression were enhanced in the presence of SFCM. SP enriched the expression of integrin subunits α4, α5, αV, β1 and β3 whereas SFCM increased α4, α5, αV, β1 and β5 integrin subunits. We also observed increased expression of Serpin E1 following SP and SFCM treatment. Wound healing scratch assay revealed enhanced migration of epithelial cells following the addition of SFCM. Taken together, we conclude that SFCM-mediated sustained activation of ZEB1, Slug in combination with upregulated migration-associated integrins and ERK (Extracellular signal-regulated kinase)-FAK-paxillin axis, may lead to induce type 2 EMT-like changes during corneal epithelial wound healing.
1. Introduction
Corneal stromal keratocytes are neural crest-derived, quiescent, mesenchymal cells of the stroma and are capable of transforming into repair-phenotype of activated stromal fibroblasts (SFs) following injury [1]. Bi-directional communication between epithelial-stromal cells plays a crucial part in the event of corneal tissue repair [2] in which epithelial cells were stimulated to proliferate by mitogenic factors produced by SFs [3]. Stromal keratocytes produce hepatocyte and keratinocyte growth factors that are in turn induce proliferation, differentiation, and motility of corneal epithelial cells by paracrine mechanisms, which are also subsequently the main processes that occur during corneal tissue repair [3,4,5]. During the course of wound healing, cultured stromal keratocytes acquire characteristics similar to those of in vivo activated myofibroblasts [6]. Additionally, conditioned media (CM) collected from SFs (SFCM) has been shown to contain epidermal, basic fibroblast growth factors and contribute to the activation of corneal epithelial cells [3,7,8]. Furthermore, activated SFs also contribute to nerve regeneration following injury. Proteomic analysis of the SFCM from activated SFs reported more than 130 proteins which are predicted to regulate focal adhesion, nerve and tissue regeneration [9].
Sensory neuropeptide substance P (SP) plays an indispensable role during wound recovery phase by functioning as an injury-inducible messenger [10,11]. SP is expressed in the corneal epithelial cells and keratocytes along with its neurokinin receptor 1 [12] and is well known to participate in the mitigation of epithelial and stromal lesions [13,14,15]. SP is also one of the several proteins that are present in the SFCM [9]. Increased SP levels were observed in cultured trigeminal neurons after treatment with corneal epithelial conditioned media [16] and also in keratocytes following injury [13]. SP enhances motility of different cell types [13,17,18] and is known to activate mitogen-activated protein kinases (MAPKs), phosphoinositide 3-kinase-Akt, protein kinase C and EGFR signaling pathways [14,19].
Epithelial-to-mesenchymal transition (EMT) is a biological process that allows polarized, stationary epithelial cells to adopt a mesenchymal cell phenotype with increased migratory capacity through a series of biochemical changes. The EMT that occurs during the repair-associated events of tissue regeneration has been classified as type 2 EMT [20,21] and is also involved in the corneal healing process [22,23]. Integrins, by interacting with extracellular matrix (ECM) components, can facilitate interactions between cells and also transduce signals that influence cell shape, proliferation, adhesion, stress fiber formation and motility [24,25]. Furthermore, they are also known to involve in a variety of inside-out and outside-in signaling events during the process of EMT [26,27,28] and corneal repair mechanisms [29,30,31,32].
The purpose of the present study was to study the effect of activated SFs on corneal epithelial function in the context of wound repair. Primary SFs were cultured and the collected SFCM was added to corneal epithelial cells in culture. Simultaneously, SP was also used to study its effect on corneal epithelial cells. Antibody microarrays were employed to profile differentially expressed cell surface markers and cytokines in the corneal epithelial cells after stimulation with SFCM and SP. Furthermore, signaling studies were performed on corneal epithelial cells with emphasis to identify the roles of SP and SFCM in mediating EMT-like changes during tissue regeneration.
2. Results
2.1. Antibody Microarray Analysis of the Differentially Expressed Proteins in Telomerase-Immortalized Human Corneal Epithelial (hTCEpi) Cells
Antibody microarrays were employed to identify putative alterations in the expression pattern of CD markers and cytokines by hTCEpi cells after treatment with either SP or SFCM (Figure 1).
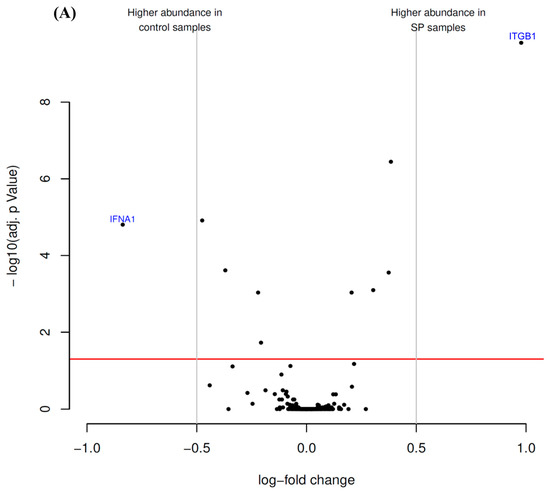
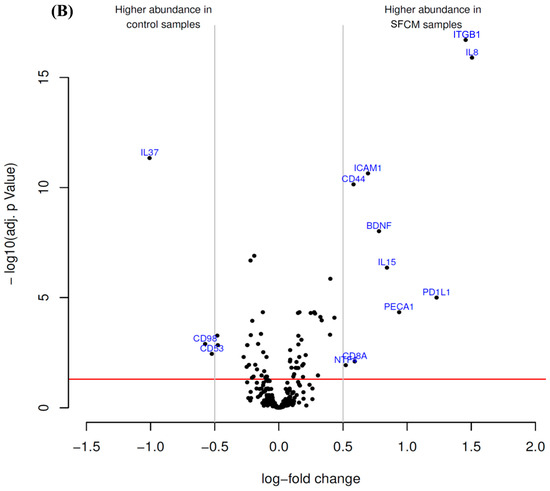
Figure 1.
Antibody microarray identified differentially enriched proteins after the treatment of hTCEpi cells with (A) substance P (SP) and (B) stromal fibroblasts conditioned medium (SFCM). The volcano plot visualizes the p-values (adjusted for multiple testing) and corresponding log-fold changes. The log-fold change of the difference in abundance is shown horizontally whereas the vertical axis indicates the significance level. The black dots represent the analyzed proteins. The horizontal red line indicates an adjusted p-value of 0.05, above which all proteins are considered to vary significantly. Proteins with a positive log-fold change had a higher abundance in SP or SFCM samples whereas proteins with a negative value in control samples.
The presence of SP in incubation medium suppressed the level of interferon-α1 (IFNA1) and potently stimulated the expression of integrin β1 (ITGB1) in comparison with control cells (Figure 1A; Table 1).

Table 1.
Proteins with most profound differential expression in hTCEpi cells after treatment with substance P (SP).
Antibody microarray analysis of hTCEpi cell lysates revealed that the treatment of cells with SFCM affected the expression of 13 proteinous products (Figure 1B). Among them, interleukin-8 (IL-8), ITGB1, programmed cell death 1 ligand 1 (PD1L1 or CD274), platelet endothelial cell adhesion molecule (PECA1 or CD31), interleukin-15 (IL-15), brain-derived neurotrophic factor (BDNF), intercellular adhesion molecule 1 (ICAM1 or CD54), T-cell surface glycoprotein CD8 α chain (CD8A), CD44, neurotrophin-4 (NTF4) were abundantly present in SFCM treated samples. In contrast, leukocyte surface antigen CD53 (CD53), 4F2 cell-surface antigen heavy chain (CD98), and interleukin-37 (IL-37) expression was suppressed in comparison with the control cells (Figure 1B, Table 2).
Table 2.
Proteins with most profound differential expression in hTCEpi cells after treatment with stromal fibroblasts conditioned media (SFCM).
The characteristics of the identified proteinous compounds with altered expression stimulated by SFCM, with respect to their subcellular localization (Table 3) and molecular function (Table 4) were obtained from STRING database. Similarly, also the lists of the biological pathways (Table 5) and processes (Table 6) were generated. The hierarchial cluster analysis data (Figure S1) and the distribution of individual array values of some proteins (Figure S2) were shown in the Supplementary Information.
Table 3.
Location of antibody microarray identified, highly enriched proteins in hTCEpi cells after treatment with SFCM.
Table 4.
Annotated molecular function of the antibody microarray identified highly abundant proteins in hTCEpi cells after treatment with SFCM.
Table 5.
Significantly impacted signaling pathways in hTCEpi cells after SFCM stimulation.
Table 6.
Significantly impacted biological processes in hTCEpi cells during SFCM stimulation.
2.2. Treatment of Corneal Epithelial Cells with SP and SFCM Activates Vital Signaling Molecules
Based on our antibody microarray results, we further intended to study activated signaling pathways upon the addition of SP and SFCM in hTCEpi cells. An increased total protein tyrosine phosphorylation in hTCEpi cells treated with SFCM was observed than with SP (Figure 2).
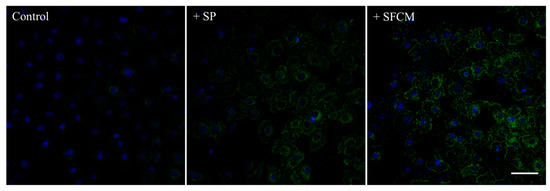
Figure 2.
Immunofluorescence detection of the total protein tyrosine phosphorylation in hTCEpi cells after treatment with SP and SFCM. Growth factor-starved hTCEpi cells were cultured in the presence of SP and SFCM for 24 h and the protein tyrosine phosphorylation was analyzed by staining with an anti-phosphotyrosine (green) antibody. An increase in the total protein tyrosine phosphorylation was observed in hTCEpi cells treated with SP and SFCM. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar: 50 μm.
Activation of MAPK family proteins within 30 min after the addition of SP and SFCM was observed. After 24 h of stimulation, increased phosphorylation of p44/42 MAPK, SAPK/JNK, and p38 kinases were observed in the presence of SP whereas phosphorylation of only p44/42 MAPK and p38 was observed in the presence of SFCM (Figure 3). Furthermore, we also analyzed the effect of SP and SFCM on the expression of important key signaling molecules related to focal adhesions, actin reorganization and EMT (Figure 4). A significant increase in the expression of paxillin was observed in the presence of SFCM. Under the both treatment conditions, either in presence of SP or SFCM, the enhanced focal adhesion kinase (FAK) and vimentin expression were observed (Figure 4).
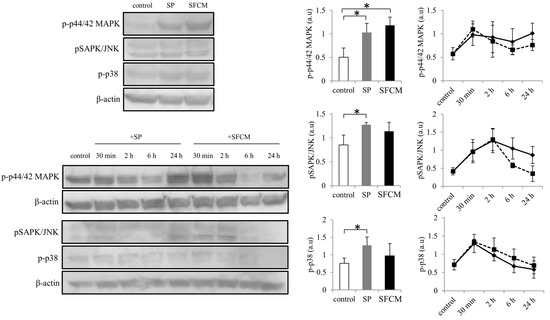
Figure 3.
Activation of mitogen-activated protein kinases (MAPK) signaling pathway following SP and SFCM treatment in hTCEpi cells. After stimulation, protein lysates were collected at time points 30 min, 2, 6, and 24 h and subjected to immunoblot analysis. Similarly, protein lysates collected directly after 24 h were also used. Phosphorylation of p44/42 MAPK, SAPK/JNK and p38 proteins were analyzed using respective antibodies. Corresponding β-actin protein levels were used to compare and calculate the differences in the phosphorylation levels. Data represent the mean of the phosphorylation levels (n > 3) shown as arbitrary units. Bar graphs indicate the mean phosphorylation levels after 24 h of treatment with SP and SFCM. In the line graphs, straight lines (―) indicate SP stimulation time points and dotted lines (…) indicate time points after SFCM stimulation. The p-values of <0.05 were considered statistically significant and are indicated by asterisks (*).
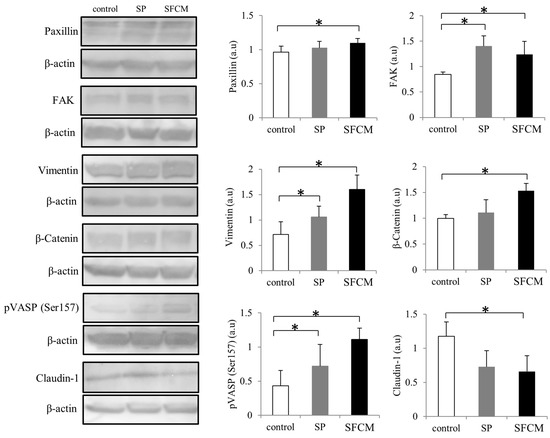
Figure 4.
Activation of important signaling molecules paxillin, focal adhesion kinase (FAK), vimentin, β-Catenin, phosphorylation of vasodilator-stimulated phosphoprotein (VASP) and claudin-1 following SP and SFCM stimulation in hTCEpi cells. Protein lysates were collected after 24 h of stimulation and subjected to immunoblot analysis. Relative expression levels of the individual proteins were analyzed using respective antibodies. Corresponding β-actin protein levels were used to compare and calculate the differences in the expression levels. Data represent the mean of the expression levels (n > 3) shown as arbitrary units. Bar graphs indicate the mean expression levels after 24 h of treatment with SP and SFCM. The p-values of <0.05 were considered statistically significant and are indicated by asterisks (*).
After the addition of SP, increased expression of FAK and vimentin were observed, whereas in case of SFCM treatment the expression of FAK was gradually increased comparing to vimentin. Levels of β-catenin were considerably enhanced in both, SP and SFCM treated cells, until 24 h whereas phosphorylated β-catenin levels were gradually decreased (Figure 5). Another important signaling molecule significantly activated in epithelial cells after the addition of SP and SFCM is vasodilator-stimulated phosphoprotein (VASP). We observed an enhanced and constitutive phosphorylation of VASP at Ser157 until 24 h (Figure 4 and Figure 5). Moreover, the presence of SP and SFCM significantly reduced the expression of tight junctional protein claudin-1 in epithelial cells (Figure 4). In addition, the expression of EMT-promoting transcription factors zinc finger E-box binding homeobox 1(ZEB1) and Slug also varied in SFCM treated cells than in SP treated cells (Figure 5). Slug expression reached its peak after 2 h of SP stimulation and reached to normal levels within 6 h. In contrast, SFCM addition leads to the increased expression levels of Slug after 2 h which was sustained until 24 h. Similar to Slug expression, SP stimulated ZEB1 expression also enhanced slightly after 2 h and reached to normal levels within 6 h whereas SFCM stimulated ZEB1 expression increased gradually until 6 h and sustained until 24 h.

Figure 5.
Differences in the expression of various important signaling molecules (ZEB1, phosphorylation of VASP, β-Catenin, integrin β1, phosphorylation of β-Catenin, integrin α4, vimentin, integrin αV, Slug and FAK) at different time points following SP and SFCM stimulation in hTCEpi cells. After stimulation, protein lysates were collected at time points 30 min, 2, 6, and 24 h and subjected to immunoblot analysis. Relative expression levels of the individual proteins were analyzed using respective antibodies. Corresponding β-actin protein levels were used to compare and calculate the differences in the expression levels. Data represent the mean of the expression levels (n > 3) shown as arbitrary units. In the line graphs, straight lines (―) indicate SP stimulation time points and dotted lines (…) indicate time points after SFCM stimulation.
2.3. Activation of Integrin Signaling
ITGB1 is the only molecule that was found to be abundantly and commonly expressed in corneal epithelial cells after the treatment with either SP or SFCM during antibody microarrays. To further understand the role of other integrins in corneal wound healing, we studied differences in the expression of various integrins (Figure 5 and Figure 6). In the presence of SP, we observed a significant increase in the expression of α4, α5, αV, β1 and β3 subunits (Figure 6). Similarly, SFCM also enhanced the expression of integrin subunits α4, α5, αV, β1 and β5 (Figure 6). Integrin β1 expression was reached its maximum after 2 h of the addition of SP and SFCM to the epithelial cells (Figure 5). Even though its expression decreased gradually, after 24 h its levels were still higher than the control. Integrin α4 expression was gradually and slightly increased during SP treatment, whereas SFCM stimulated increase in α4 integrin reached its maximum levels in 2 h and was persistent until 24 h (Figure 5 and Figure 6).
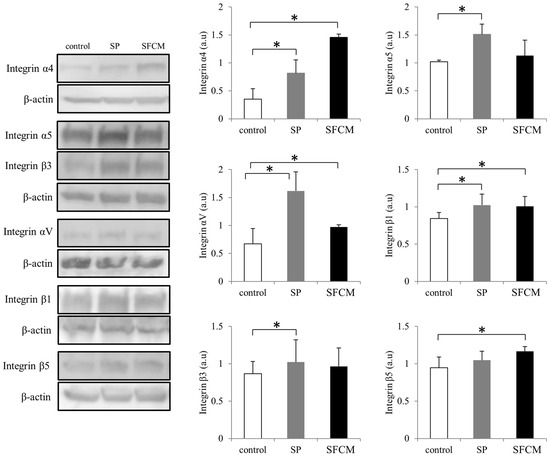
Figure 6.
Differences in the expression of various integrin subunits (α4, α5, αV, β1, β3 and β5) following SP and SFCM stimulation in hTCEpi cells. Protein lysates were collected after 24 h of stimulation and subjected to immunoblot analysis. Relative expression levels of the individual proteins were analyzed using respective antibodies. Corresponding β-actin protein levels were used to compare and calculate the differences in the expression levels. Data represent the mean of the expression levels (n > 3) shown as arbitrary units. Bar graphs indicate the mean expression levels after 24 h of treatment with SP and SFCM. The p-values of <0.05 were considered statistically significant and are indicated by asterisks (*).
We also observed considerably enhanced CD44 expression in our cultured epithelial cells following the addition of SFCM (Figure 7). Additionally, increased expression of Serpin E1, another important injury-response molecule, was also observed in the presence of SP and SFCM (Figure 7).
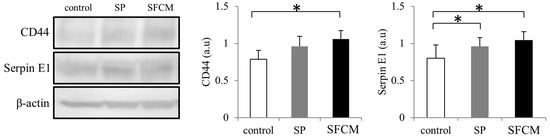
Figure 7.
Differences in the expression of CD44 and Serpin E1 following SP and SFCM stimulation in hTCEpi cells. Protein lysates were collected after 24 h of stimulation and subjected to immunoblot analysis. Relative expression levels of the individual proteins were analyzed using respective antibodies. Corresponding β-actin protein levels were used to compare and calculate the differences in the expression levels. Data represent the mean of the expression levels (n > 3) shown as arbitrary units. Bar graphs indicate the mean expression levels after 24 h of treatment with SP and SFCM. The p-values of <0.05 were considered statistically significant and are indicated by asterisks (*).
2.4. SFCM Enhances Epithelial Cell Motility and Migration
To further study the role of SP and SFCM in corneal epithelial cell motility and migration, we performed a scratch assay. The presence of SFCM enhances motility and migration of corneal epithelial cells, in comparison to the control and SP treated cells (Figure 8). During wound healing assay, SFCM treated corneal epithelial cells filled the scratch area much faster than untreated control and SP treated cells. Cell migration was represented as a number of cells filling the central gap area after making the scratch. We observed a significant increase in the number of migrating cells in the presence of SFCM in both conditions, 12 h as well as 24 h, compared to the control and SP treated cells after making the scratch (Figure 8). We believe that addition of SFCM increases also cell proliferation [8] along with motility and migration in comparison to the SP or untreated control cells.
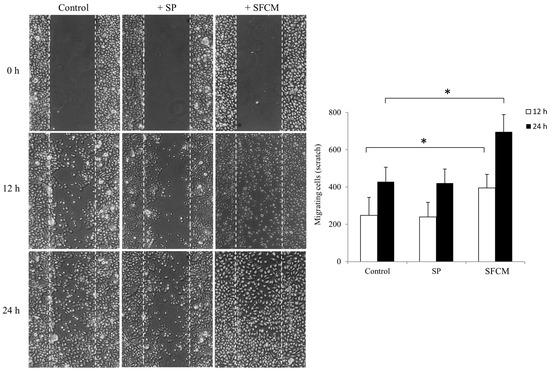
Figure 8.
Wound healing scratch assay was made on 24 h growth factor-starved, confluent hTCEpi cells by scratching a line across the bottom of the culture dish. SP and SFCM were added to the culture media and the cell motility and migration was observed at time points 12 h and 24 h, respectively. The micrographs show the extent of scratch closure obtained under control conditions compared to those with the addition of SP and SFCM. Cell migration quantification was evaluated by counting the number of cells in the central gap. Three independent experiments were performed and a representative result is shown. The p-values of <0.05 were considered statistically significant and are indicated by asterisks (*).
3. Discussion
Epithelial-mesenchymal cell communications are essential to regulate wounded tissue regeneration following injury [33,34]. Similarly, in the cornea, cell–cell interactions play dominant role throughout the process of wound healing [35], in which cytokine, growth factor and chemokine-mediated stromal-epithelial interactions are indispensable [2,3,5,36,37,38,39,40]. Additionally, conditioned media from the corneal stromal cells is also known to stimulate epithelial cell growth, proliferation and nerve regeneration due to the presence of various cytokines, keratins, growth factors, chemokines, neurotrophic factors and vital signaling molecules [7,8,9,41]. The present work was focused on to unravel the functional molecules that are necessary to promote cell growth, motility and proliferation of wounded corneal epithelial cells. Furthermore, we explored this aspect, in the context of epithelial-stromal interactions, using antibody microarrays to identify differentially regulated cell surface markers and cytokines that are essential for the epithelial cell function. Since SFCM contains surplus essential factors, including neuropeptide protachykinin-1, than stromal keratocyte conditioned medium [9], we employed CM collected from the SFs alongside SP in the present study to investigate their trophic effects on corneal epithelial cells.
Antibody microarray analysis revealed a higher abundance of ITGB1 in SP treated cells, which plays an important role in cell-ECM adhesion, migration, tissue repair [42,43] and its appearance in the cornea was correlated with the process of corneal epithelial wound repair [29]. Treatment of epithelial cells with SFCM resulted to the differential expression of several proteins that have either direct or indirect roles in epithelial cell growth, movement, and adhesion-related signaling cascades during tissue regeneration. Similar to the SP treatment, we also observed upregulated ITGB1 in SFCM treated corneal epithelial cells. IL-8 contributes to cell migration and chemotaxis during tissue repair [44,45,46]. IL-8 release from the cells is mainly regulated at the transcriptional level and MAPK pathway plays an important role in stabilizing IL-8 mRNA and its protein expression [47,48]. Since trophic factors in the CM also regulate IL-8 synthesis [49], the observed increase in IL-8 production after the addition of SFCM may be due to the increased stability of IL-8 mRNA facilitated in part by the trophic factors in SFCM together with SFCM-mediated MAPK pathway activation. CD274 modulates the corneal immune response and functions as an anti-angiogenic molecule [50,51] and its up-regulation after the addition of SFCM may contribute to protect immune privilege status of the wounded cornea during tissue renewal. Cell surface molecule CD31 participates in governing cell adhesion and activation of key signal transduction cascades by recruiting adaptor proteins [52,53]. Pro-inflammatory cytokine IL-15 promotes cell proliferation, migration, and regeneration in response to injury [54,55,56] and also stimulates the production of IL-8 [57]. The collective production of IL-15 and IL-8 following the addition of SFCM may induce EMT-like changes [58,59] to augment epithelial cell migration and tissue regeneration following injury. CD54 is upregulated in the healing corneas and participate in the epithelial recruitment of T cells along with enhanced cell motility and migration [60,61,62]. Cells lacking T-cell receptor also express CD8A and its expression is known to be modulated by cytokines [63,64,65]. CD44 is a predominant cell surface glycoprotein receptor and adhesion molecule which plays an important role in signal transduction processes related to cell adhesion, wound healing, EMT [66,67,68,69] and its expression correlates with re-epithelialization in the healing cornea [70]. The nerve growth factor family of neurotrophins including BDNF and NTF4 mediate trophic properties and are highly expressed during corneal nerve regeneration [71,72,73]. They promote neurite outgrowth and necessary for the survival of axotomized neurons [74,75,76] as well as growth and proliferation of non-neuronal cells by activation of MAPK signaling pathway [77,78,79]. SFCM induced expression of BDNF, NT4 may also promote epithelial cell proliferation in conjunction with corneal nerve regeneration after injury and further emphasize the interdependence of stromal-epithelial cell alliance in corneal nerve regeneration. Protein–protein interaction analysis, following the treatment of corneal epithelial cells with SFCM, revealed distinct direct as well as indirectly interconnected networks among the abundantly expressed cell surface molecules and cytokines (Figure S3), additionally highlights the prominence of stromal–epithelial interactions during corneal epithelial tissue repair.
Coordinated collective cell migration through sustained intercellular connections is the fundamental cell movement during corneal epithelial wound repair in vivo [80,81,82] where adhesive cell-substratum interactions, including cell–cell and cell–ECM adhesions, are mandatory for sustained cell motility and migration towards the injured site [36,83,84]. During in vitro scratch-wound healing models, lamellipodial crawling is the most commonly observed migration mechanism where each polarized cell progress through different structural alterations that include lamellipodium formation, nucleus translocation and trailing edge detachment [82,85]. This mechanism is further regulated by complex events of actin polymerization, depolymerization, and integrin signaling [86,87]. Correspondingly, alteration of plasticity plays an indispensable part in the process of EMT during which cells lose their epithelial characteristics without impairing the cell-cell interactions associated with collective migration and acquire mesenchymal qualities associated with individual migration in a reversible manner [88,89,90]. This transition further involves synchronized alterations in various additional cell structures including actin cytoskeleton reorganization, engagement, and expression of various integrins which navigate cells through the ECM [88,91,92]. During the course of tissue repair, all these coordinated processes are in part executed by complex and overlapping signaling networks involving growth factors, cytokines, and chemokines [93].
Reorganization of the cytoskeleton along with actin filament remodeling at the leading edge is essential for EMT and cell migration during epithelial injury. VASP mediated focal adhesion, actin filament binding and polymerization inhibits cell movement whereas its phosphorylation inhibits actin polymerization [94] and thereby induces epithelial cell motility during repair along with alteration of various protein–protein interactions [95,96]. The observed consistent phosphorylation of VASP at Ser157, but not at Ser239, in corneal epithelial cells in the presence of SFCM and SP may assist in locating VASP at the leading edge to induce filopodia formation [97] during cell motility. Additionally, phosphorylated VASP mediated actin remodeling events dismantle cell–cell junctions during the progression of EMT [98,99]. During cell migration, protein tyrosine kinase FAK acts downstream of different growth factor and ECM components and is a central player of integrin-mediated motility [100]. Furthermore, it activates focal adhesion signaling proteins and regulates cell adhesion disassembly through adopter protein paxillin [101]. In our study, after stimulation of cells with SP and SFCM, we also observed increased expression of FAK, paxillin along with steady activation of p44/42 MAPK and integrin β1. Since ERK (Extracellular signal-regulated kinase)-FAK-paxillin pathway regulates cell migration along with the recruitment of integrin β1 [102,103], we also presume the activation of ERK-FAK-paxillin-integrin β1 cascade in the presence of SP and SFCM during corneal epithelial wound repair. Vimentin functions as a signal integrator during EMT, wound repair and also regulates structure and function of focal adhesions by linking its intermediate filaments to paxillin [104,105,106,107]. Epithelial vimentin expression during the corneal wound repair is linked to cell–matrix interactions and cell migration [108]. In addition, vimentin also binds to p44/42 MAPK and prolongs its activation by preventing dephosphorylation [109]. The vimentin-p44/42 MAPK axis further regulates the transcriptional activity of Slug to establish and promote EMT-like alterations [110]. As the expression of vimentin, p44/42 MAPK correlates with transcription factor Slug expression in the course of EMT, their consistent activation in the presence of SFCM may also indicate the incidence of EMT-like modifications during epithelial tissue repair.
Integrins are heterodimeric proteins composed of transmembrane α, β subunits and are important for epithelial cell migration during corneal tissue regeneration [31]. Integrin expression is regulated by cytokines and growth factors [111]. Integrin β1expression is enhanced during corneal wound repair in actively migrating epithelial cells [29], and its deletion leads to unstable adhesions and delayed wound healing [112]. Integrin subunits αV, β1 and β5 mediate interactions with ECM proteins and migration [31,113] whereas integrin α4 promotes motility and serves as EMT marker protein [114,115] and α5 triggers β-catenin mediated EMT and migration [116]. Integrin αV and β3 contributes to the TGF-β mediated EMT process through FAK and ERK-mediated pathways by dissociating cell–cell junctions [27,28,117]. Furthermore, SP-induced effects were mediated by αV, β3 subunits [118] and SP enhances α5 expression [119]. Similarly, in this study, we also observed increased expression of α5, αV, as well as β3 integrins confirming SP facilitated influence on epithelial cells. SFCM facilitated upregulation of migration-associated α4, α5, αV, β1 and β5 integrins may play a role in inducing the migration of epithelial cells as we observed in our scratch assay. Furthermore, by modulating the function of integrins and ECM, SerpinE1 also induce cell migration along with EMT during wound healing [120,121]. Since vimentin binding to integrin cytoplasmic tails regulate integrin-ligand interactions [122] which in turn propagates through MAPK pathway [123,124], the observed enhanced expression of EMT markers, migration-associated integrins in conjunction with activated ERK-FAK-paxillin axis may have an influence on SFCM mediated cell proliferation and migration in this study.
The zinc finger transcription factors Slug and ZEB1 regulate cell migration and basic events of EMT [20]. Slug regulates transcriptional activation of ZEB1, controls the expression of adhesion molecules and integrins together with suppression of epithelial claudin-1 expression [125,126,127]. Similarly, ZEB1 also involved in the activation of focal adhesion formation and cytoskeletal remodeling events [128,129]. In corneal epithelial cells we also observed Slug dependent expression of ZEB1. Increased Slug levels were correlated with increased ZEB1 levels during SFCM stimulation which was lasted until 24 h. In addition, the decreased claudin-1 levels also correlate with Slug in SFCM treated epithelial cells which may sequentially lead to the onset of EMT-like transition [130]. The key player of canonical Wnt signaling, β-catenin, is also expressed in the corneal epithelium where its activation escalates epithelial cell proliferation and EMT [20,131]. Similarly, elevated β-catenin protein levels were detected during wound healing [132,133]. Non-phosphorylated β-catenin translocates to the nucleus and activates various transcription factors whereas Ser33, Ser37, and Thr41 phosphorylated form targeted for ubiquitin-mediated proteasomal degradation. As Slug is also known to promote the activation of β-catenin [134], sustained β-catenin in conjunction with activated integrins and Slug [135] may also shift the equilibrium towards cell migration and contribute to type 2 EMT-like alterations in the epithelial cells following the addition of SFCM.
SP treatment alone is insufficient to promote injury induced corneal epithelial cell migration [136,137] and SP-mediated effects are mediated in synergy with other growth factors [136,137,138,139] during corneal wound healing. In accordance with these observations, we also did not observe any SP-mediated cell migration during our scratch assay. Instead, the SP-mediated activation of MAPK signaling pathway, integrins, and other key signaling molecules that we observed in our study may contribute to the corneal epithelial sensitization, homeostasis and wound repair, in synergy with other growth factors [139,140,141]. Furthermore, the enhanced proliferation and migration of epithelial cells upon the addition of SFCM correlates with the presence of numerous amounts of trophic factors in the stromal conditioned medium [9]. Taken together, SFCM mediated enhanced and sustained activation of ZEB1, Slug transcription factors in combination with upregulated migration-associated integrins and ERK-FAK-paxillin axis, may lead to induce type 2 EMT-like changes during wound healing. Based on our results, we conclude that stromal-epithelial interactions, by activating various interconnected cell surface molecules, growth factors and cytokines, may control key intracellular signaling events that are crucial to promote EMT-like changes along with nerve regeneration during tissue repair at the ocular surface.
4. Materials and Methods
4.1. Materials
The following antibodies used in the present study were purchased from Cell Signaling Technology (Frankfurt, Germany); Phospho-tyrosine (Cat no. 9411), phospho-SAPK/JNK (Cat no. 4668), phospho-p38 MAPK (Cat no. 4511), phospho-p44/42 MAPK (Cat no. 4370), claudin-1 (Cat no. 13255), TCF8/ZEB1 (Cat no. 3396), phospho-VASP (Ser157) (Cat no. 3111), vimentin (Cat no. 5741), β-Catenin (Cat no. 8480), phospho-β-Catenin (Ser33/37/Thr41) (Cat no. 9561), Slug (Cat no. 9585), FAK (Cat no. 3285), paxillin (Cat no. 12065), integrin α4 (Cat no. 8440), integrin α5 (Cat no. 4705), integrin αV (Cat no. 4711), integrin β1 (Cat no. 9699), integrin β3 (Cat no. 13166), integrin β5 (Cat no. 3629). CD44 (Cat no. MA5-13890) was from Thermo Fischer (Darmstadt, Germany), Serpin E1 (Cat no. MAB1786) was purchased from R&D systems (Wiesbaden-Nordenstadt, Germany) and β-actin (Cat no. A5316) antibody was from Sigma-Aldrich (Munich, Germany). Anti-rabbit IgG- HRP antibody (Cat no. 7074) and anti-mouse IgG-HRP antibody (Cat no. 7076) were purchased from Cell Signaling Technology (Frankfurt, Germany). TPP tissue culture flasks, dishes and 6, 12-well plates were obtained from Sigma-Aldrich. Coverslips and glass slides were from Marienfeld (Bonn, Germany). Enhanced chemiluminescence (ECL) prime detection western blotting reagent was purchased from Amersham (Munich, Germany) and other western blotting reagents were from Bio-Rad (Munich, Germany). Substance P (SP) (Cat no. 1156) was purchased from Tocris (Wiesbaden-Nordenstadt, Germany).
4.2. Cell Culture
This study was approved by the ethics committee of the University of Rostock (Approved ID: A 2014-0100) and followed the guidelines of the Declaration of Helsinki.
In this present study, hTCEpi cell-line [142], between passages 20–28, was used (a kind gift of James V Jester; authenticated and characterized according to ATCC standard protocols). hTCEpi cells were cultured in KGM-Gold™ growth medium (Lonza, Köln, Germany). Cells were sub-cultured on T75 tissue culture flasks (Sigma-Aldrich), incubated at 37 °C in 5% CO2 and passaged every 5 to 7 days.
Primary human SFs were cultured using an explant culture method, after collecting corneas from donor cadavers, in DMEM with low glucose (Sigma-Aldrich) supplemented with 10% fetal calf serum. SFs were seen growing out of the corneal explants after 3–4 days. When outgrowing primary SFs reached a confluent monolayer, cells were trypsinized and subcultured. For experiments in this present study, SFs of third passage were used. The mesenchymal origin of the cells was confirmed by immune staining with antibody against vimentin.
For the collection of CM from the cultured primary SFs, KGM-Gold medium without any supplemented growth factors was added to the phosphate buffered salinePBS washed confluent cultures and the respective SFCM was collected after 24 h to stimulate hTCEpi cells immediately (3:1 dilution with normal KGM-Gold medium without supplemented growth factors) or stored at −70 °C. The collected SFCM was spun at 300× g for 5 min to remove any remaining cell debris. During stimulation of hTCEpi cells, confluent cultures were growth factor-starved for 24 h before stimulation and SP was added at the concentration of 10−5 M along with the growth factor-deprived cell culture media.
4.3. Antibody Microarray Analysis
To analyze the differential expression of CD markers and cytokines (scio CD—Cell surface marker and Cytokine profiling) in hTCEpi cells in the presence of SFCM and SP, cells were treated with SFCM and SP, for 24 h as described above. Later, the cells were collected, washed and frozen cell pellets were sent to Sciomics GmbH (Heidelberg, Germany) for further analysis. For each condition, the array was performed in triplicates. Briefly, proteins were extracted, quantified and labeled with fluorescent dyes. All nine samples were analyzed in a dual-color approach using a reference-based design on scioCD antibody microarrays (Sciomics) targeting 95 different CD surface markers and 26 cytokines/chemokines with 270 different monoclonal antibodies. The list of the analyzed target proteins was provided as supplementary information. Each antibody is represented in eight replicates on the array. The arrays were blocked with scioBlock (Sciomics) on a Hybstation 4800 (Tecan, Grödig, Austria). Resulting data were analyzed using the linear models for microarray data (LIMMA) package of R-Bioconductor after uploading the median signal intensities for differential protein expression. For normalization, a specialized invariant Lowess method was applied and for analysis of the samples, a one-factorial linear model was fitted with LIMMA resulting in a two-sided t-test or F-test based on moderated statistics. All presented p values were adjusted for multiple testing by controlling the false discovery rate according to Benjamini and Hochberg. Proteins were defined as differential for |logFC| > 0.5 and an adjusted p-value < 0.05. Differences in protein abundance between different samples or sample groups are presented as log-fold changes (logFC) calculated for the basis 2. In a study comparing samples versus control a logFC = 1 means that the sample group had on average a 21 = 2-fold higher signal as the control group. logFC = −1 stands for 2−1 = 1/2 of the signal in the sample as compared to the control group.
4.4. Immunofluorescence
Epithelial cells grown on glass coverslips were fixed with paraformaldehyde (4%, w/v in PBS) for 10 min after washing with PBS and then permeabilized with PBS containing 0.1% Triton X-100 (Sigma-Aldrich) for 30 min. Fixed and permeabilized cells were then incubated with primary antibody for intracellular staining. Primary antibody in this study was used at 1:100 dilution in PBS containing 0.1% Triton X-100 with 2% FCS and incubated for 60 min at room temperature. The cells were then washed with PBS before adding secondary donkey anti-mouse IgG (H + L)-Alexa Fluor 488 (diluted 1:500) and incubated at room temperature for another 60 min. Later, cells were washed 3 times with PBS and mounted in mounting medium (Vector Labs, Eching, Germany) containing 4,6-diamidino-2-phenylindole (DAPI). Cells were observed under a Nikon confocal fluorescence microscope equipped with a digital camera (Nikon Eclipse E400 with D-Eclipse C1) (Dusseldorf, Germany) and all images were taken from a single plane through the cell monolayers with 40× objective using the same settings.
4.5. Immunoblotting
After treatment with SFCM or SP, hTCEpi cell monolayers were washed with PBS and lysed in RIPA buffer (Sigma-Aldrich) containing protease and phosphatase inhibitors (Roche, Mannheim, Germany). Equal amounts of total cell lysates were loaded into the wells of 10% Mini-PROTEAN® TGX™ precast gels (Bio-Rad), separated by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad). Later, membranes were blocked with 5% non-fat dry milk (Carl Roth, Karlsruhe, Germany) in Tris-buffered saline with 0.1% Tween-20 (TBS-T) for 30 min and incubated with respective primary antibodies (diluted 1:1000) at 4 °C overnight. After washing 3 times with TBS-T, membranes were incubated with secondary HRP-conjugated anti-rabbit or anti-mouse IgG (diluted 1:2500) for an additional 1 h at room temperature and developed to visualize protein bands using the ECL detection system. During quantification, the optical density of each protein band was normalized to the corresponding β-actin band. Quantification of the blots was performed using ImageJ software [143].
4.6. Scratch Wound Healing Assay
Migration of wounded corneal epithelial cells was assessed by performing an in vitro scratch assay in which a linear wound midline was made across the bottom of the dish on a confluent monolayer of epithelial cells using a 200 µL sterile pipet tip. After that, cells were rinsed gently with PBS to remove any remaining cell debris. KGM-Gold™ medium deprived of growth factors was used during the scratch assay. SFCM and SP were used as mentioned in Section 2.2. Micrographs of the cells were taken at 10× magnification using a microscope equipped with a Moticam10 digital camera at time points 0, 12 and 24 h. Epithelial cell migration across the wound line was then quantified by counting the cells invading the central scratch area in the control, SP and SFCM treated culture dishes.
4.7. Statistical Analyses
Bar charts and line plots were generated using means and the standard deviation. Student’s t-test was used for the comparison between two groups (control vs. SP; control vs. SFCM) and p-values of <0.05 were considered statistically significant and are indicated by asterisks.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/19/2/464/s1.
Acknowledgments
The authors thank Colette Leyh for her technical skills and expertise. We also thank James V Jester, University of California, USA for providing immortalized hTCEpi cells. This work was financially supported by Deutsche Forschungsgemeinschaft (DFG) (KO-4979/1-1).
Author Contributions
Bhavani S. Kowtharapu conceived and designed the experiments, performed the experiments, analyzed the data and wrote the paper; Radovan Murin analyzed the data and wrote the paper; Anselm G. M. Jünemann and Oliver Stachs participated in the design and coordination of the study. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| BDNF | Brain-derived neurotrophic factor |
| CD | Cluster of differentiation |
| CD8A | T-cell surface glycoprotein CD8 α chain |
| CD98 | 4F2 cell-surface antigen heavy chain |
| CM | Conditioned media |
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| FAK | Focal adhesion kinase |
| hTCEpi | Telomerase-immortalized human corneal epithelial cell line |
| ICAM1 | Intercellular adhesion molecule 1 |
| IFNA1 | Interferon-α1 |
| ITGB1 | Integrin β1 |
| IL-8 | Interleukin-8 |
| IL-15 | Interleukin-15 |
| IL-37 | Interleukin-37 |
| LIMMA | linear models for microarray data |
| MAPK | Mitogen-activated protein kinases |
| NTF4 | Neurotrophin-4 |
| PECA1 | Platelet endothelial cell adhesion molecule |
| PD1L1 | Programmed cell death 1 ligand 1 |
| SFs | Stromal fibroblasts |
| SFCM | Stromal fibroblasts conditioned media |
| SP | Substance P |
| VASP | Vasodilator-stimulated phosphoprotein |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
References
- West-Mays, J.A.; Dwivedi, D.J. The keratocyte: Corneal stromal cell with variable repair phenotypes. Int. J. Biochem. Cell Biol. 2006, 38, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Liu, J.J.; Mohan, R.R. Stromal-epithelial interactions in the cornea. Prog. Retin. Eye Res. 1999, 18, 293–309. [Google Scholar] [CrossRef]
- Jacob, T.J.; Cruwys, S.; Mackie, I.A. Control of cellular proliferation in the bovine cornea: An in vitro study. Eye 1989, 3 Pt 5, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; He, Y.G.; Weng, J.; Zieske, J.D.; Jester, J.V.; Schultz, G.S. Effect of epidermal growth factor, hepatocyte growth factor, and keratinocyte growth factor, on proliferation, motility and differentiation of human corneal epithelial cells. Exp. Eye Res. 1994, 59, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Sotozono, C.; Kinoshita, S.; Kita, M.; Imanishi, J. Paracrine role of keratinocyte growth factor in rabbit corneal epithelial cell growth. Exp. Eye Res. 1994, 59, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Jester, J.V.; Barry, P.A.; Lind, G.J.; Petroll, W.M.; Garana, R.; Cavanagh, H.D. Corneal keratocytes: In situ and in vitro organization of cytoskeletal contractile proteins. Investig. Ophthalmol. Vis. Sci. 1994, 35, 730–743. [Google Scholar]
- Chan, K.Y.; Haschke, R.H. Epithelial-stromal interactions: Specific stimulation of corneal epithelial cell growth in vitro by a factor(s) from cultured stromal fibroblasts. Exp. Eye Res. 1983, 36, 231–246. [Google Scholar] [CrossRef]
- Pancholi, S.; Tullo, A.; Khaliq, A.; Foreman, D.; Boulton, M. The effects of growth factors and conditioned media on the proliferation of human corneal epithelial cells and keratocytes. Graefes Arch. Clin. Exp. Ophthalmol. 1998, 236, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yam, G.H.; Williams, G.P.; Setiawan, M.; Yusoff, N.Z.; Lee, X.W.; Htoon, H.M.; Zhou, L.; Fuest, M.; Mehta, J.S. Nerve regeneration by human corneal stromal keratocytes and stromal fibroblasts. Sci. Rep. 2017, 7, 45396. [Google Scholar] [CrossRef] [PubMed]
- Brain, S.D. Sensory neuropeptides: Their role in inflammation and wound healing. Immunopharmacology 1997, 37, 133–152. [Google Scholar] [CrossRef]
- Hong, H.S.; Lee, J.; Lee, E.; Kwon, Y.S.; Lee, E.; Ahn, W.; Jiang, M.H.; Kim, J.C.; Son, Y. A new role of substance P as an injury-inducible messenger for mobilization of CD29+ stromal-like cells. Nat. Med. 2009, 15, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakayasu, K.; Iwatsu, M.; Kanai, A. Endogenous substance P in corneal epithelial cells and keratocytes. Jpn. J. Ophthalmol. 2002, 46, 616–620. [Google Scholar] [CrossRef]
- Sloniecka, M.; Le Roux, S.; Zhou, Q.; Danielson, P. Substance P Enhances Keratocyte Migration and Neutrophil Recruitment through Interleukin-8. Mol. Pharmacol. 2016, 89, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Di, G.; Qi, X.; Qu, M.; Wang, Y.; Duan, H.; Danielson, P.; Xie, L.; Zhou, Q. Substance P promotes diabetic corneal epithelial wound healing through molecular mechanisms mediated via the neurokinin-1 receptor. Diabetes 2014, 63, 4262–4274. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kawahara, M.; Nakata, K.; Nishida, T. Restoration of corneal epithelial barrier function and wound healing by substance P and IGF-1 in rats with capsaicin-induced neurotrophic keratopathy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2937–2940. [Google Scholar] [CrossRef]
- Kowtharapu, B.S.; Stahnke, T.; Wree, A.; Guthoff, R.F.; Stachs, O. Corneal epithelial and neuronal interactions: Role in wound healing. Exp. Eye Res. 2014, 125, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ruff, M.R.; Wahl, S.M.; Pert, C.B. Substance P receptor-mediated chemotaxis of human monocytes. Peptides 1985, 6 (Suppl. 2), 107–111. [Google Scholar] [CrossRef]
- Chernova, I.; Lai, J.P.; Li, H.; Schwartz, L.; Tuluc, F.; Korchak, H.M.; Douglas, S.D.; Kilpatrick, L.E. Substance P (SP) enhances CCL5-induced chemotaxis and intracellular signaling in human monocytes, which express the truncated neurokinin-1 receptor (NK1R). J. Leukoc. Biol. 2009, 85, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ramnath, R.D.; Tamizhselvi, R.; Bhatia, M. Role of protein kinase C and phosphoinositide 3-kinase-Akt in substance P-induced proinflammatory pathways in mouse macrophages. FASEB J. 2009, 23, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Rigaud, M. Epithelial Mesenchymal Transition: A double-edged sword. Clin. Transl. Med. 2015, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Aomatsu, K.; Arao, T.; Sugioka, K.; Matsumoto, K.; Tamura, D.; Kudo, K.; Kaneda, H.; Tanaka, K.; Fujita, Y.; Shimomura, Y.; Nishio, K. TGF-β induces sustained upregulation of SNAI1 and SNAI2 through Smad and non-Smad pathways in a human corneal epithelial cell line. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Aomatsu, K.; Arao, T.; Abe, K.; Kodama, A.; Sugioka, K.; Matsumoto, K.; Kudo, K.; Kimura, H.; Fujita, Y.; Hayashi, H.; et al. Slug is upregulated during wound healing and regulates cellular phenotypes in corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Byun, J.S.; Gardner, K. Wounds that will not heal: Pervasive cellular reprogramming in cancer. Am. J. Pathol. 2013, 182, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Gervasi, M.E.; Bakin, A. Role of β5-integrin in epithelial-mesenchymal transition in response to TGF-β. Cell Cycle 2010, 9, 1647–1659. [Google Scholar] [CrossRef] [PubMed]
- Mamuya, F.A.; Wang, Y.; Roop, V.H.; Scheiblin, D.A.; Zajac, J.C.; Duncan, M.K. The roles of αV integrins in lens EMT and posterior capsular opacification. J. Cell. Mol. Med. 2014, 18, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.; Nishida, T.; Otori, T. Coordinated appearance of β 1 integrins and fibronectin during corneal wound healing. J. Lab. Clin. Med. 1992, 120, 86–93. [Google Scholar] [PubMed]
- Stepp, M.A. Corneal integrins and their functions. Exp. Eye Res. 2006, 83, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.T. The role of integrins in corneal wound healing. Vet. Ophthalmol. 2009, 12 (Suppl. 1), 2–9. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Mezquita, J.T.; Hutcheon, A.E.; Stepp, M.A.; Zieske, J.D. αVβ6 integrin promotes corneal wound healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8505–8513. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.C.; Tan, M.J.; Philippe, V.; Tan, S.H.; Tan, C.K.; Ku, C.W.; Goh, Y.Y.; Wahli, W.; Michalik, L.; Tan, N.S. Regulation of epithelial-mesenchymal IL-1 signaling by PPARβ/Δ is essential for skin homeostasis and wound healing. J. Cell Biol. 2009, 184, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Netto, M.; Ambrosio, R., Jr. Corneal cells: Chatty in development, homeostasis, wound healing, and disease. Am. J. Ophthalmol. 2003, 136, 530–536. [Google Scholar] [CrossRef]
- Suzuki, K.; Saito, J.; Yanai, R.; Yamada, N.; Chikama, T.; Seki, K.; Nishida, T. Cell-matrix and cell-cell interactions during corneal epithelial wound healing. Prog. Retin. Eye Res. 2003, 22, 113–133. [Google Scholar] [CrossRef]
- Lim, M.; Goldstein, M.H.; Tuli, S.; Schultz, G.S. Growth factor, cytokine and protease interactions during corneal wound healing. Ocul. Surf. 2003, 1, 53–65. [Google Scholar] [CrossRef]
- Kakazu, A.; Chandrasekher, G.; Bazan, H.E. HGF protects corneal epithelial cells from apoptosis by the PI-3K/Akt-1/Bad- but not the ERK1/2-mediated signaling pathway. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3485–3492. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Wang, D.Y.; Kao, M.H.; Chen, J.K. The growth-promoting effect of KGF on limbal epithelial cells is mediated by upregulation of ΔNp63α through the p38 pathway. J. Cell Sci. 2009, 122, 4473–4480. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kobayashi, T.; Shiraishi, A.; Hara, Y.; Kadota, Y.; Yang, L.; Inoue, T.; Shirakata, Y.; Ohashi, Y. Stromal-epithelial interaction study: The effect of corneal epithelial cells on growth factor expression in stromal cells using organotypic culture model. Exp. Eye Res. 2015, 135, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Amirjamshidi, H.; Milani, B.Y.; Sagha, H.M.; Movahedan, A.; Shafiq, M.A.; Lavker, R.M.; Yue, B.Y.; Djalilian, A.R. Limbal fibroblast conditioned media: A non-invasive treatment for limbal stem cell deficiency. Mol. Vis. 2011, 17, 658–666. [Google Scholar] [PubMed]
- Liu, S.; Xu, S.W.; Blumbach, K.; Eastwood, M.; Denton, C.P.; Eckes, B.; Krieg, T.; Abraham, D.J.; Leask, A. Expression of integrin β1 by fibroblasts is required for tissue repair in vivo. J. Cell Sci. 2010, 123, 3674–3682. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.R.; Hilgarth, R.S.; Leoni, G.; Kwon, M.; Den Beste, K.A.; Parkos, C.A.; Nusrat, A. Annexin A2 regulates β1 integrin internalization and intestinal epithelial cell migration. J. Biol. Chem. 2013, 288, 15229–15239. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Sanders, A.J.; Ruge, F.; Harding, K.G. Influence of interleukin-8 (IL-8) and IL-8 receptors on the migration of human keratinocytes, the role of PLC-γ and potential clinical implications. Exp. Ther. Med. 2012, 3, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Rennekampff, H.O.; Hansbrough, J.F.; Kiessig, V.; Dore, C.; Sticherling, M.; Schroder, J.M. Bioactive interleukin-8 is expressed in wounds and enhances wound healing. J. Surg. Res. 2000, 93, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Eslani, M.; Movahedan, A.; Afsharkhamseh, N.; Sroussi, H.; Djalilian, A.R. The role of toll-like receptor 4 in corneal epithelial wound healing. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6108–6115. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.; Lausch, R.N.; Oakes, J.E. Substance P differentially stimulates IL-8 synthesis in human corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3871–3877. [Google Scholar]
- Bhattacharyya, S.; Gutti, U.; Mercado, J.; Moore, C.; Pollard, H.B.; Biswas, R. MAPK signaling pathways regulate IL-8 mRNA stability and IL-8 protein expression in cystic fibrosis lung epithelial cell lines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L81–L87. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.C.; Maa, M.C.; Tsai, F.J.; Chen, W.C.; Lin, J.G.; Jeng, L.B.; Yang, R.S.; Fu, W.M.; Tang, C.H. Osteoblast-derived TGF-β1 stimulates IL-8 release through AP-1 and NF-kappaB in human cancer cells. J. Bone Min. Res. 2008, 23, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Bukowiecki, A.; Hos, D.; Cursiefen, C.; Eming, S.A. Wound-Healing Studies in Cornea and Skin: Parallels, Differences and Opportunities. Int. J. Mol. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chauhan, S.K.; El Annan, J.; Sage, P.T.; Sharpe, A.H.; Dana, R. A novel function for programmed death ligand-1 regulation of angiogenesis. Am. J. Pathol. 2011, 178, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.A.; Yan, H.C.; Madri, J.A.; Albelda, S.M. Platelet endothelial cell adhesion molecule, PECAM-1, modulates cell migration. J. Cell. Physiol. 1992, 153, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Ilan, N.; Cheung, L.; Pinter, E.; Madri, J.A. Platelet-endothelial cell adhesion molecule-1 (CD31), a scaffolding molecule for selected catenin family members whose binding is mediated by different tyrosine and serine/threonine phosphorylation. J. Biol. Chem. 2000, 275, 21435–21443. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; McCall, S.; Choi, S.S.; Sicklick, J.K.; Huang, J.; Qi, Y.; Zdanowicz, M.; Camp, T.; Li, Y.X.; Diehl, A.M. Interleukin-15 increases hepatic regenerative activity. J. Hepatol. 2006, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Kagimoto, Y.; Yamada, H.; Ishikawa, T.; Maeda, N.; Goshima, F.; Nishiyama, Y.; Furue, M.; Yoshikai, Y. A regulatory role of interleukin 15 in wound healing and mucosal infection in mice. J. Leukoc. Biol. 2008, 83, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Griffiths, J.L.; Sanders, A.J.; Owen, S.; Ruge, F.; Harding, K.G.; Jiang, W.G. The clinical significance and impact of interleukin 15 on keratinocyte cell growth and migration. Int. J. Mol. Med. 2016, 38, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Ponzi, A.N.; Millesimo, M.; Notarangelo, L.D.; Musso, T. Interleukin-15 (IL-15) induces IL-8 and monocyte chemotactic protein 1 production in human monocytes. Blood 1997, 90, 2804–2809. [Google Scholar] [PubMed]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Meng, X.; Guo, W.; Cai, P.; Li, W.; Li, Q.; Wang, W.; Sun, Y.; Xu, Q.; Gu, Y. Transmembrane-Bound IL-15-Promoted Epithelial-Mesenchymal Transition in Renal Cancer Cells Requires the Src-Dependent Akt/GSK-3β/β-Catenin Pathway. Neoplasia 2015, 17, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wu, H.K.; Bruce, A.; Wollenberg, K.; Panjwani, N. Detection of differentially expressed genes in healing mouse corneas, using cDNA microarrays. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2897–2904. [Google Scholar]
- Byeseda, S.E.; Burns, A.R.; Dieffenbaugher, S.; Rumbaut, R.E.; Smith, C.W.; Li, Z. ICAM-1 is necessary for epithelial recruitment of γδ T cells and efficient corneal wound healing. Am. J. Pathol. 2009, 175, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Fushimi, N.; Suzuki, Y.; Suzuki, M.; Sakimoto, T.; Sawa, M. Intercellular adhesion molecule-1 expression on human corneal epithelial outgrowth from limbal explant in culture. Br. J. Ophthalmol. 2003, 87, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Adoro, S.; Lucas, P.J.; Sarafova, S.D.; Alag, A.S.; Doan, L.L.; Erman, B.; Liu, X.; Ellmeier, W.; Bosselut, R.; et al. ‘Coreceptor tuning’: Cytokine signals transcriptionally tailor CD8 coreceptor expression to the self-specificity of the TCR. Nat. Immunol. 2007, 8, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, D.J.; Marcet-Palacios, M.; Sekar, Y.; Ng, M.C.; Befus, A.D. CD8 α is expressed by human monocytes and enhances FcγR-dependent responses. BMC Immunol. 2007, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.H.; Baz, A.; Groves, P.; Kelso, A.; Kienzle, N. Interferon-γ and interleukin-4 reciprocally regulate CD8 expression in CD8+ T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 17475–17480. [Google Scholar] [CrossRef] [PubMed]
- Asari, A.; Morita, M.; Sekiguchi, T.; Okamura, K.; Horie, K.; Miyauchi, S. Hyaluronan, CD44 and fibronectin in rabbit corneal epithelial wound healing. Jpn. J. Ophthalmol. 1996, 40, 18–25. [Google Scholar] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Griffin, C.T.; Wang, S.S.; Bissell, D.M. Role of CD44 in epithelial wound repair: Migration of rat hepatic stellate cells utilizes hyaluronic acid and CD44v6. J. Biol. Chem. 2005, 280, 15398–15404. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Park, Y.S.; Kim, H.J.; Kim, C.H.; Lim, S.W.; Huh, J.W.; Lee, J.H.; Kim, H.R. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int. J. Oncol. 2012, 41, 211–218. [Google Scholar] [PubMed]
- Yu, F.X.; Guo, J.; Zhang, Q. Expression and distribution of adhesion molecule CD44 in healing corneal epithelia. Investig. Ophthalmol. Vis. Sci. 1998, 39, 710–717. [Google Scholar]
- Barbacid, M. Neurotrophic factors and their receptors. Curr. Opin. Cell Biol. 1995, 7, 148–155. [Google Scholar] [CrossRef]
- Chaudhary, S.; Namavari, A.; Yco, L.; Chang, J.H.; Sonawane, S.; Khanolkar, V.; Sarkar, J.; Jain, S. Neurotrophins and nerve regeneration-associated genes are expressed in the cornea after lamellar flap surgery. Cornea 2012, 31, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Kruse, F.E.; Volcker, H.E. Neurotrophic factors in the human cornea. Investig. Ophthalmol. Vis. Sci. 2000, 41, 692–702. [Google Scholar]
- Ibanez, C.F.; Ernfors, P.; Timmusk, T.; Ip, N.Y.; Arenas, E.; Yancopoulos, G.D.; Persson, H. Neurotrophin-4 is a target-derived neurotrophic factor for neurons of the trigeminal ganglion. Development 1993, 117, 1345–1353. [Google Scholar] [PubMed]
- Cohen, A.; Bray, G.M.; Aguayo, A.J. Neurotrophin-4/5 (NT-4/5) increases adult rat retinal ganglion cell survival and neurite outgrowth in vitro. J. Neurobiol. 1994, 25, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Peinado-Ramon, P.; Salvador, M.; Villegas-Perez, M.P.; Vidal-Sanz, M. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Investig. Ophthalmol. Vis. Sci. 1996, 37, 489–500. [Google Scholar]
- Yoshizaki, K.; Yamamoto, S.; Yamada, A.; Yuasa, K.; Iwamoto, T.; Fukumoto, E.; Harada, H.; Saito, M.; Nakasima, A.; Nonaka, K.; et al. Neurotrophic factor neurotrophin-4 regulates ameloblastin expression via full-length TrkB. J. Biol. Chem. 2008, 283, 3385–3391. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Fujita, T.; Kajiya, M.; Takeda, K.; Shiba, H.; Kawaguchi, H.; Kurihara, H. Brain-derived neurotrophic factor induces migration of endothelial cells through a TrkB-ERK-integrin αVβ3-FAK cascade. J. Cell. Physiol. 2012, 227, 2123–2129. [Google Scholar] [CrossRef] [PubMed]
- Revest, J.M.; Le Roux, A.; Roullot-Lacarriere, V.; Kaouane, N.; Vallee, M.; Kasanetz, F.; Rouge-Pont, F.; Tronche, F.; Desmedt, A.; Piazza, P.V. BDNF-TrkB signaling through Erk1/2 MAPK phosphorylation mediates the enhancement of fear memory induced by glucocorticoids. Mol. Psychiatry 2014, 19, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Y.; Zhao, M.; Jiang, J. Collective cell migration: Implications for wound healing and cancer invasion. Burns Trauma 2013, 1, 21–26. [Google Scholar] [PubMed]
- Buck, R.C. Cell migration in repair of mouse corneal epithelium. Investig. Ophthalmol. Vis. Sci. 1979, 18, 767–784. [Google Scholar]
- Fong, E.; Tzlil, S.; Tirrell, D.A. Boundary crossing in epithelial wound healing. Proc. Natl. Acad. Sci. USA 2010, 107, 19302–19307. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, T. Cell adhesion and its endocytic regulation in cell migration during neural development and cancer metastasis. Int. J. Mol. Sci. 2012, 13, 4564–4590. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, L.; Heino, J.; Hakkinen, L.; Larjava, H. Integrins in Wound Healing. Adv. Wound Care (New Rochelle) 2014, 3, 762–783. [Google Scholar] [CrossRef] [PubMed]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Arciero, J.C.; Mi, Q.; Branca, M.F.; Hackam, D.J.; Swigon, D. Continuum model of collective cell migration in wound healing and colony expansion. Biophys. J. 2011, 100, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.Y.; Javaid, S.; Wong, E.A.; Perk, S.; Haber, D.A.; Toner, M.; Irimia, D. Collective and individual migration following the epithelial-mesenchymal transition. Nat. Mater. 2014, 13, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, S.; Theveneau, E.; Benedetto, A.; Parsons, M.; Tanaka, M.; Charras, G.; Kabla, A.; Mayor, R. In vivo collective cell migration requires an LPAR2-dependent increase in tissue fluidity. J. Cell Biol. 2014, 206, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 2008, 582, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Casanova, J. A common framework for EMT and collective cell migration. Development 2016, 143, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, B.; Huttelmaier, S.; Schluter, K.; Jockusch, B.M.; Illenberger, S. Phosphorylation of the vasodilator-stimulated phosphoprotein regulates its interaction with actin. J. Biol. Chem. 2000, 275, 30817–30825. [Google Scholar] [CrossRef] [PubMed]
- Hastie, A.T.; Wu, M.; Foster, G.C.; Hawkins, G.A.; Batra, V.; Rybinski, K.A.; Cirelli, R.; Zangrilli, J.G.; Peters, S.P. Alterations in vasodilator-stimulated phosphoprotein (VASP) phosphorylation: Associations with asthmatic phenotype, airway inflammation and β2-agonist use. Respir. Res. 2006, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Benz, P.M.; Blume, C.; Seifert, S.; Wilhelm, S.; Waschke, J.; Schuh, K.; Gertler, F.; Munzel, T.; Renne, T. Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J. Cell Sci. 2009, 122, 3954–3965. [Google Scholar] [CrossRef] [PubMed]
- Doppler, H.R.; Bastea, L.I.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z.; Storz, P. Protein kinase D1-mediated phosphorylations regulate vasodilator-stimulated phosphoprotein (VASP) localization and cell migration. J. Biol. Chem. 2013, 288, 24382–24393. [Google Scholar] [CrossRef] [PubMed]
- Sperry, R.B.; Bishop, N.H.; Bramwell, J.J.; Brodeur, M.N.; Carter, M.J.; Fowler, B.T.; Lewis, Z.B.; Maxfield, S.D.; Staley, D.M.; Vellinga, R.M.; et al. Zyxin controls migration in epithelial-mesenchymal transition by mediating actin-membrane linkages at cell-cell junctions. J. Cell. Physiol. 2010, 222, 612–624. [Google Scholar] [PubMed]
- Kim, G.T.; Lee, S.H.; Kim, Y.M. Torilis japonica extract, a new potential EMT suppressor agent by regulation of EGFR signaling pathways. Int. J. Oncol. 2014, 45, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Crowe, D.L.; Ohannessian, A. Recruitment of focal adhesion kinase and paxillin to β1 integrin promotes cancer cell migration via mitogen activated protein kinase activation. BMC Cancer 2004, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Teranishi, S.; Kimura, K.; Nishida, T. Role of formation of an ERK-FAK-paxillin complex in migration of human corneal epithelial cells during wound closure in vitro. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5646–5652. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J. Vimentin: Central hub in EMT induction? Small GTPases 2011, 2, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindstrom, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-β-Slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef] [PubMed]
- Menko, A.S.; Bleaken, B.M.; Libowitz, A.A.; Zhang, L.; Stepp, M.A.; Walker, J.L. A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol. Biol. Cell 2014, 25, 776–790. [Google Scholar] [CrossRef] [PubMed]
- SundarRaj, N.; Rizzo, J.D.; Anderson, S.C.; Gesiotto, J.P. Expression of vimentin by rabbit corneal epithelial cells during wound repair. Cell Tissue Res. 1992, 267, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Perlson, E.; Michaelevski, I.; Kowalsman, N.; Ben-Yaakov, K.; Shaked, M.; Seger, R.; Eisenstein, M.; Fainzilber, M. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J. Mol. Biol. 2006, 364, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Virtakoivu, R.; Mai, A.; Mattila, E.; de Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin-ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.T.; Yamada, K.M. The regulation of expression of integrin receptors. Proc. Soc. Exp. Biol. Med. 1997, 214, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Grose, R.; Hutter, C.; Bloch, W.; Thorey, I.; Watt, F.M.; Fassler, R.; Brakebusch, C.; Werner, S. A crucial role of β 1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 2002, 129, 2303–2315. [Google Scholar] [PubMed]
- Varadarajulu, J.; Laser, M.; Hupp, M.; Wu, R.; Hauck, C.R. Targeting of α(v) integrins interferes with FAK activation and smooth muscle cell migration and invasion. Biochem. Biophys. Res. Commun. 2005, 331, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Young, S.A.; McCabe, K.E.; Bartakova, A.; Delaney, J.; Pizzo, D.P.; Newbury, R.O.; Varner, J.A.; Schlaepfer, D.D.; Stupack, D.G. Integrin α4 Enhances Metastasis and May Be Associated with Poor Prognosis in MYCN-low Neuroblastoma. PLoS ONE 2015, 10, e0120815. [Google Scholar] [CrossRef] [PubMed]
- Konac, E.; Kiliccioglu, I.; Sogutdelen, E.; Dikmen, A.U.; Albayrak, G.; Bilen, C.Y. Do the expressions of epithelial-mesenchymal transition proteins, periostin, integrin-α4 and fibronectin correlate with clinico-pathological features and prognosis of metastatic castration-resistant prostate cancer? Exp. Biol. Med. (Maywood) 2017, 242, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Renner, G.; Noulet, F.; Mercier, M.C.; Choulier, L.; Etienne-Selloum, N.; Gies, J.P.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. Expression/activation of α5β1 integrin is linked to the β-catenin signaling pathway to drive migration in glioma cells. Oncotarget 2016, 7, 62194–62207. [Google Scholar] [CrossRef] [PubMed]
- Pin, A.L.; Huot, J. β5 integrin orchestrates epithelial mesenchymal transition in breast cancer: Comment on: Bianchi A, et al. Cell Cycle 2010, 9, 1647–1659. Cell Cycle 2010, 9, 1873–1877. [Google Scholar] [CrossRef]
- Koon, H.W.; Zhao, D.; Zhan, Y.; Moyer, M.P.; Pothoulakis, C. Substance P mediates antiapoptotic responses in human colonocytes by Akt activation. Proc. Natl. Acad. Sci. USA 2007, 104, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Chikama, T.; Nishida, T. Up-regulation of integrin α 5 expression by combination of substance P and insulin-like growth factor-1 in rabbit corneal epithelial cells. Biochem. Biophys. Res. Commun. 1998, 246, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Higgins, C.E.; Czekay, R.P.; Law, B.K.; Higgins, S.P.; Archambeault, J.; Kutz, S.M.; Higgins, P.J. SERPINE1: A Molecular Switch in the Proliferation-Migration Dichotomy in Wound-“Activated” Keratinocytes. Adv. Wound Care (New Rochelle) 2014, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Longmate, W.M.; Law, B.K.; Higgins, P.J. Targeted Inhibition of PAI-1 Activity Impairs Epithelial Migration and Wound Closure Following Cutaneous Injury. Adv. Wound Care (New Rochelle) 2015, 4, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, C.; Kim, E.J.; Jang, J.; Kim, S.J.; Kang, S.M.; Kim, M.G.; Jung, H.; Park, D.; Kim, C. Vimentin filaments regulate integrin-ligand interactions by binding to the cytoplasmic tail of integrin β3. J. Cell Sci. 2016, 129, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.F.; Chaudhary, L.; Fausto, A.; Halstead, L.R.; Ory, D.S.; Avioli, L.V.; Cheng, S.L. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J. Biol. Chem. 2001, 276, 14443–14450. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.L.; Weaver, V.M.; Hammer, D.A. Integrin-mediated signalling through the MAP-kinase pathway. IET Syst. Biol. 2008, 2, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wels, C.; Joshi, S.; Koefinger, P.; Bergler, H.; Schaider, H. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J. Investig. Dermatol. 2011, 131, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Turner, F.E.; Broad, S.; Khanim, F.L.; Jeanes, A.; Talma, S.; Hughes, S.; Tselepis, C.; Hotchin, N.A. Slug regulates integrin expression and cell proliferation in human epidermal keratinocytes. J. Biol. Chem. 2006, 281, 21321–21331. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Estrada, O.M.; Culleres, A.; Soriano, F.X.; Peinado, H.; Bolos, V.; Martinez, F.O.; Reina, M.; Cano, A.; Fabre, M.; Vilaro, S. The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem. J. 2006, 394, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Gibbons, D.L.; Chakravarti, D.; Creighton, C.J.; Rizvi, Z.H.; Adams, H.P.; Pertsemlidis, A.; Gregory, P.A.; Wright, J.A.; Goodall, G.J.; et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Investig. 2012, 122, 3170–3183. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Takano, K.; Yamamoto, T.; Murata, M.; Son, S.; Imamura, M.; Yamaguchi, H.; Osanai, M.; Chiba, H.; Himi, T.; et al. Transforming growth factor-β induces epithelial to mesenchymal transition by down-regulation of claudin-1 expression and the fence function in adult rat hepatocytes. Liver Int. 2008, 28, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, M.N.; Ding, Z.; Ng, M.Y.; Truong, T.T.; Yu, F.; Deng, S.X. Wnt/β-catenin signaling regulates proliferation of human cornea epithelial stem/progenitor cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4734–4741. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.S.; Cheah, A.Y.; Turley, S.; Nadesan, P.; Poon, R.; Clevers, H.; Alman, B.A. β-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc. Natl. Acad. Sci. USA 2002, 99, 6973–6978. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.S.; Wei, Q.; Gurung, A.; Youn, A.; Bright, T.; Poon, R.; Whetstone, H.; Guha, A.; Alman, B.A. Β-catenin regulates wound size and mediates the effect of TGF-β in cutaneous healing. FASEB J. 2006, 20, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug promote epithelial-mesenchymal transition through β-catenin-T-cell factor-4-dependent expression of transforming growth factor-β3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.M.; Gurski, L.A.; Engel, C.; Gnarra, J.R.; Maranchie, J.K.; Pilch, J. Integrin αvβ3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer Res. 2013, 73, 6175–6184. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Nakamura, M.; Ofuji, K.; Reid, T.W.; Mannis, M.J.; Murphy, C.J. Synergistic effects of substance P with insulin-like growth factor-1 on epithelial migration of the cornea. J. Cell. Physiol. 1996, 169, 159–166. [Google Scholar] [CrossRef]
- Kingsley, R.E.; Marfurt, C.F. Topical substance P and corneal epithelial wound closure in the rabbit. Investig. Ophthalmol. Vis. Sci. 1997, 38, 388–395. [Google Scholar]
- Nakamura, M.; Chikama, T.; Nishida, T. Synergistic effect with Phe-Gly-Leu-Met-NH2 of the C-terminal of substance P and insulin-like growth factor-1 on epithelial wound healing of rabbit cornea. Br. J. Pharmacol. 1999, 127, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Suvas, S. Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J. Immunol. 2017, 199, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Yanai, R.; Inui, M.; Nishida, T. Sensitizing effect of substance P on corneal epithelial migration induced by IGF-1, fibronectin, or interleukin-6. Investig. Ophthalmol. Vis. Sci. 2005, 46, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, F.; di Zazzo, A.; de Simone, L.; Bonini, S. The Intriguing Role of Neuropeptides at the Ocular Surface. Ocul. Surf. 2017, 15, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.M.; Li, L.; Fisher, S.; Pearce, V.P.; Shay, J.W.; Wright, W.E.; Cavanagh, H.D.; Jester, J.V. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Investig. Ophthalmol. Vis. Sci. 2005, 46, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Grenacher, B.; Rohde, B.; Vogel, J. Quantifying Western blots: Pitfalls of densitometry. Electrophoresis 2009, 30, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).