Common Variable Immunodeficiency and Gastric Malignancies



Abstract
1. Introduction
2. Genetic Abnormalities
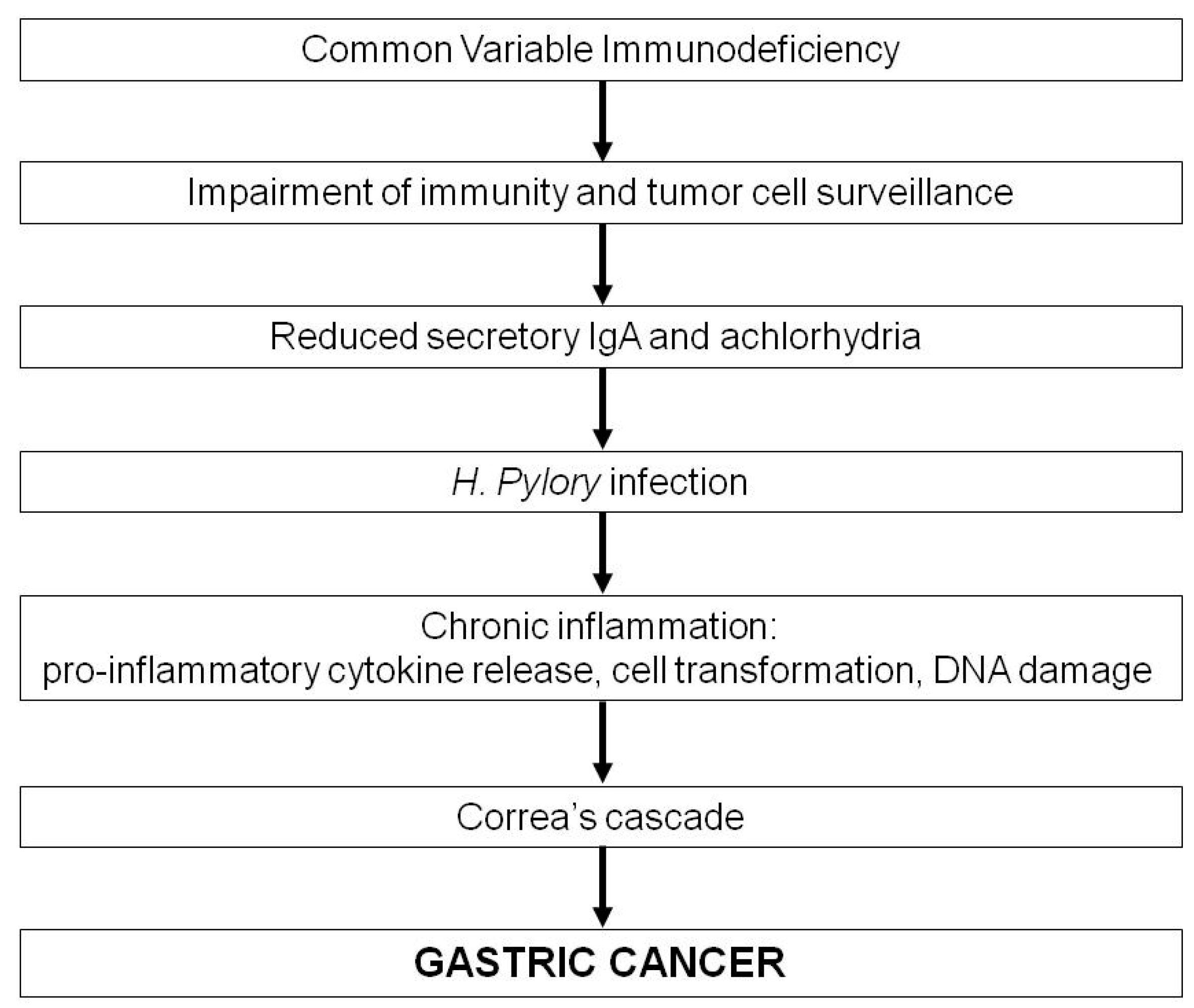
3. Common Variable Immunodeficiency and Gastric Cancer
4. Features of CVID-Associated Gastric Cancer
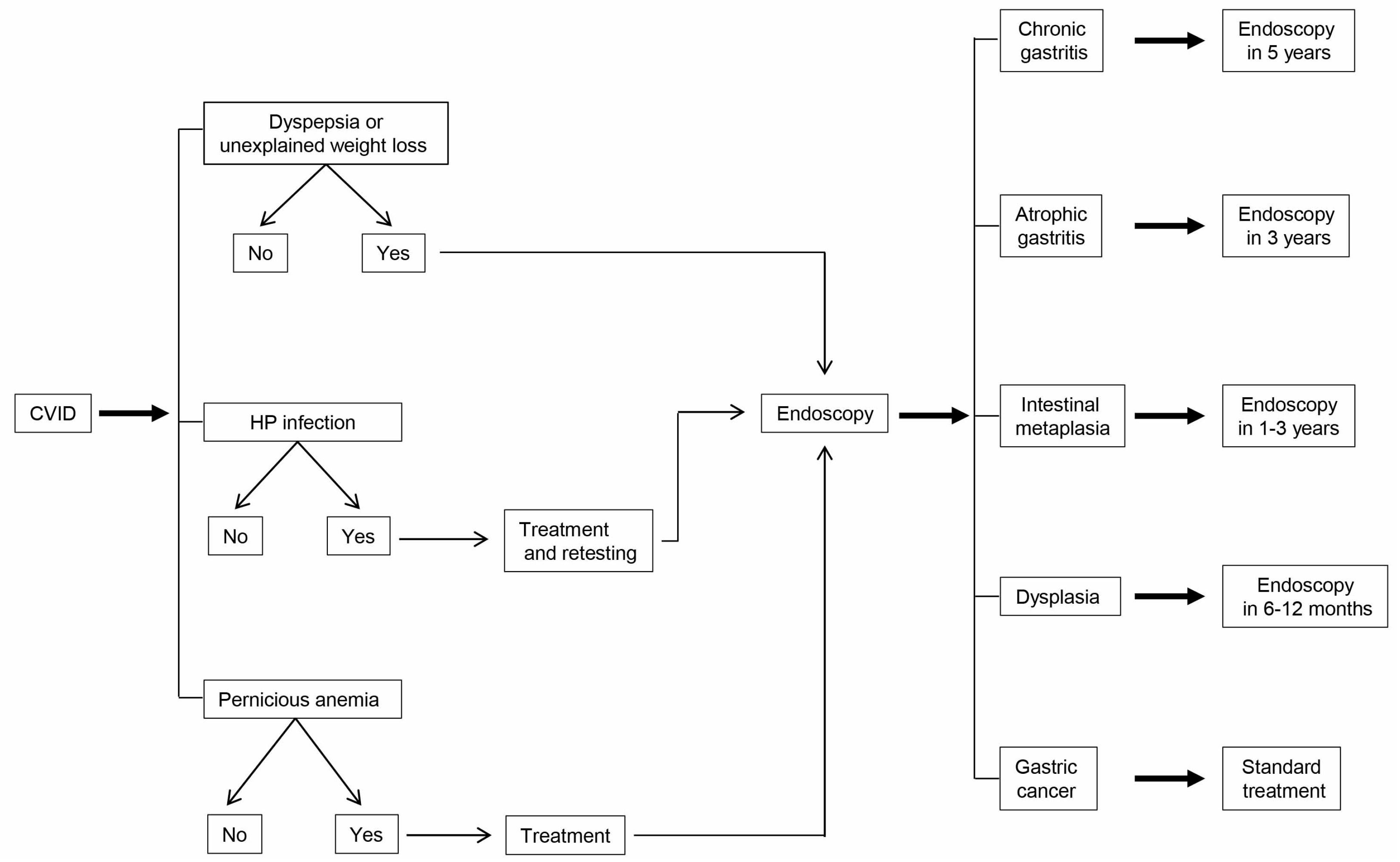
5. Gastric Cancer Screening and Prevention in CVID Patients
6. CVID and Gastric Lymphoma
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abbott, J.K.; Gelfand, E.W. Common Variable Immunodeficiency: Diagnosis, Management, and Treatment. Immunol. Allergy Clin. N. Am. 2015, 35, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Anzilotti, C.; Kienzler, A.K.; Lopez-Granados, E.; Gooding, S.; Davies, B.; Pandit, H.; Lucas, M.; Price, A.; Littlewood, T.; van der Burg, M.; et al. Key stages of bone marrow B-cell maturation are defective in patients with common variable immunodeficiency disorders. J. Allergy Clin. Immunol. 2015, 136, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Warnatz, K.; Denz, A.; Drager, R.; Braun, M.; Groth, C.; Wolff-Vorbeck, G.; Eibel, H.; Schlesier, M.; Peter, H.H. Severe deficiency of switched memory B cells (CD27+IgM−IgD−) in subgroups of patients with common variable immunodeficiency: A new approach to classify a heterogeneous disease. Blood 2002, 99, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Arandi, N.; Mirshafiey, A.; Jeddi-Tehrani, M.; Abolhassani, H.; Sadeghi, B.; Mirminachi, B.; Shaghaghi, M.; Aghamohammadi, A. Evaluation of CD4+CD25+FOXP3+ regulatory T cells function in patients with common variable immunodeficiency. Cell. Immunol. 2013, 281, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.; Zhou, Z.; George, I.; Becker, K.; Cunningham-Rundles, C. Enhanced apoptosis of T cells in common variable immunodeficiency (CVID): Role of defective CD28 co-stimulation. Clin. Exp. Immunol. 2000, 120, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, A.; Pierdominici, M.; Mazzetta, F.; Marziali, M.; Renzi, C.; Mileo, A.M.; de Felice, M.; Mora, B.; Esposito, A.; Carello, R.; et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J. Immunol. 2007, 178, 3932–3943. [Google Scholar] [CrossRef] [PubMed]
- Taraldsrud, E.; Fevang, B.; Aukrust, P.; Beiske, K.H.; Floisand, Y.; Froland, S.; Rollag, H.; Olweus, J. Common variable immunodeficiency revisited: Normal generation of naturally occurring dendritic cells that respond to Toll-like receptors 7 and 9. Clin. Exp. Immunol. 2014, 175, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Viallard, J.F.; Camou, F.; Andre, M.; Liferman, F.; Moreau, J.F.; Pellegrin, J.L.; Blanco, P. Altered dendritic cell distribution in patients with common variable immunodeficiency. Arthritis Res. Ther. 2005, 7, R1052–R1055. [Google Scholar] [CrossRef] [PubMed]
- Ochtrop, M.L.; Goldacker, S.; May, A.M.; Rizzi, M.; Draeger, R.; Hauschke, D.; Stehfest, C.; Warnatz, K.; Goebel, H.; Technau-Ihling, K.; et al. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood 2011, 118, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Conley, M.E.; Notarangelo, L.D.; Etzioni, A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin. Immunol. 1999, 93, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B.; Fieschi, C.; Thon, V.; Abedi, M.R.; Hammarstrom, L. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood 2008, 112, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Barlan, I.; Chapel, H.; Costa-Carvalho, B.T.; Cunningham-Rundles, C.; de la Morena, M.T.; Espinosa-Rosales, F.J.; Hammarstrom, L.; Nonoyama, S.; Quinti, I.; et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract. 2016, 4, 38–59. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, M.; Ko, H.M.; Mehandru, S.; Cunningham-Rundles, C. Gastrointestinal Disorders Associated with Common Variable Immune Deficiency (CVID) and Chronic Granulomatous Disease (CGD). Curr. Gastroenterol. Rep. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Rundles, C.; Cooper, D.L.; Duffy, T.P.; Strauchen, J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am. J. Hematol. 2002, 69, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gathmann, B.; Mahlaoui, N.; Gerard, L.; Oksenhendler, E.; Warnatz, K.; Schulze, I.; Kindle, G.; Kuijpers, T.W.; Dutch, W.I.D.; et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Vajdic, C.M.; Mao, L.; van Leeuwen, M.T.; Kirkpatrick, P.; Grulich, A.E.; Riminton, S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood 2010, 116, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Kinlen, L.J.; Webster, A.D.; Bird, A.G.; Haile, R.; Peto, J.; Soothill, J.F.; Thompson, R.A. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet 1985, 1, 263–266. [Google Scholar] [CrossRef]
- Castigli, E.; Wilson, S.A.; Garibyan, L.; Rachid, R.; Bonilla, F.; Schneider, L.; Geha, R.S. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet. 2005, 37, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Bacchelli, C.; Buckridge, S.; Pan-Hammarstrom, Q.; Jennings, S.; Lougaris, V.; Bergbreiter, A.; Hagena, T.; Birmelin, J.; Plebani, A.; et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 2009, 113, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Mackay, C.R.; Mackay, F. The BAFF/APRIL system: Life beyond B lymphocytes. Mol. Immunol. 2005, 42, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Castigli, E.; Scott, S.; Dedeoglu, F.; Bryce, P.; Jabara, H.; Bhan, A.K.; Mizoguchi, E.; Geha, R.S. Impaired IgA class switching in APRIL-deficient mice. Proc. Natl. Acad. Sci. USA 2004, 101, 3903–3908. [Google Scholar] [CrossRef] [PubMed]
- Romberg, N.; Virdee, M.; Chamberlain, N.; Oe, T.; Schickel, J.N.; Perkins, T.; Cantaert, T.; Rachid, R.; Rosengren, S.; Palazzo, R.; et al. TNF receptor superfamily member 13b (TNFRSF13B) hemizygosity reveals transmembrane activator and CAML interactor haploinsufficiency at later stages of B-cell development. J. Allergy Clin. Immunol. 2015, 136, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Abolhassani, H.; Kiaee, F.; Tavakolinia, N.; Rafiemanesh, H.; Yazdani, R.; Mahdaviani, S.A.; Mohammadikhajehdehi, S.; Tavakol, M.; Ziaee, V.; et al. Autoimmunity and its association with regulatory T cells and B cell subsets in patients with common variable immunodeficiency. Allergol. Immunopathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pan-Hammarstrom, Q.; Salzer, U.; Du, L.; Bjorkander, J.; Cunningham-Rundles, C.; Nelson, D.L.; Bacchelli, C.; Gaspar, H.B.; Offer, S.; Behrens, T.W.; et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 2007, 39, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Pieper, K.; Rizzi, M.; Speletas, M.; Smulski, C.R.; Sic, H.; Kraus, H.; Salzer, U.; Fiala, G.J.; Schamel, W.W.; Lougaris, V.; et al. A common single nucleotide polymorphism impairs B-cell activating factor receptor’s multimerization, contributing to common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 133, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Schweighoffer, E.; Vanes, L.; Nys, J.; Cantrell, D.; McCleary, S.; Smithers, N.; Tybulewicz, V.L. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 2013, 38, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Castigli, E.; Wilson, S.A.; Scott, S.; Dedeoglu, F.; Xu, S.; Lam, K.P.; Bram, R.J.; Jabara, H.; Geha, R.S. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 2005, 201, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.A.; Dillon, S.R.; Mudri, S.; Johnston, J.; Littau, A.; Roque, R.; Rixon, M.; Schou, O.; Foley, K.P.; Haugen, H.; et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. impaired B cell maturation in mice lacking BLyS. Immunity 2001, 15, 289–302. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Bende, R.J.; Baars, P.A.; Grummels, A.; Derks, I.A.; Dolman, K.M.; Beaumont, T.; Tedder, T.F.; van Noesel, C.J.; Eldering, E.; et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J. Clin. Investig. 2010, 120, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Thiel, J.; Kimmig, L.; Salzer, U.; Grudzien, M.; Lebrecht, D.; Hagena, T.; Draeger, R.; Voelxen, N.; Bergbreiter, A.; Jennings, S.; et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J. Allergy Clin. Immunol. 2012, 129, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Van Zelm, M.C.; Smet, J.; Adams, B.; Mascart, F.; Schandene, L.; Janssen, F.; Ferster, A.; Kuo, C.C.; Levy, S.; van Dongen, J.J.; et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Investig. 2010, 120, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Maul-Pavicic, A.; Cunningham-Rundles, C.; Urschel, S.; Belohradsky, B.H.; Litzman, J.; Holm, A.; Franco, J.L.; Plebani, A.; Hammarstrom, L.; et al. ICOS deficiency in patients with common variable immunodeficiency. Clin. Immunol. 2004, 113, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Oh, E.; Ng, C.H.; Lam, K.P. Impaired germinal center formation and recall T-cell-dependent immune responses in mice lacking the costimulatory ligand B7-H2. Blood 2003, 102, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Offer, S.M.; Pan-Hammarstrom, Q.; Hammarstrom, L.; Harris, R.S. Unique DNA repair gene variations and potential associations with the primary antibody deficiency syndromes IgAD and CVID. PLoS ONE 2010, 5, e12260. [Google Scholar] [CrossRef] [PubMed]
- Tampella, G.; Baronio, M.; Vitali, M.; Soresina, A.; Badolato, R.; Giliani, S.; Plebani, A.; Lougaris, V. Evaluation of CARMA1/CARD11 and Bob1 as candidate genes in common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2011, 21, 348–353. [Google Scholar] [PubMed]
- Orange, J.S.; Glessner, J.T.; Resnick, E.; Sullivan, K.E.; Lucas, M.; Ferry, B.; Kim, C.E.; Hou, C.; Wang, F.; Chiavacci, R.; et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J. Allergy Clin. Immunol. 2011, 127, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Bode, C.; Kenefeck, R.; Hou, T.Z.; Wing, J.B.; Kennedy, A.; Bulashevska, A.; Petersen, B.S.; Schaffer, A.A.; Gruning, B.A.; et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 2014, 20, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Deau, M.C.; Heurtier, L.; Frange, P.; Suarez, F.; Bole-Feysot, C.; Nitschke, P.; Cavazzana, M.; Picard, C.; Durandy, A.; Fischer, A.; et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J. Clin. Investig. 2014, 124, 3923–3928. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Coonrod, E.M.; Kumanovics, A.; Franks, Z.F.; Durtschi, J.D.; Margraf, R.L.; Wu, W.; Heikal, N.M.; Augustine, N.H.; Ridge, P.G.; et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 2013, 93, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.J.; Remmers, E.F.; Sun, G.; Freeman, A.F.; Datta, S.; Torabi-Parizi, P.; Subramanian, N.; Bunney, T.D.; Baxendale, R.W.; Martins, M.S.; et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N. Engl. J. Med. 2012, 366, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Herrera, G.; Tampella, G.; Pan-Hammarstrom, Q.; Herholz, P.; Trujillo-Vargas, C.M.; Phadwal, K.; Simon, A.K.; Moutschen, M.; Etzioni, A.; Mory, A.; et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am. J. Hum. Genet. 2012, 90, 986–1001. [Google Scholar] [CrossRef] [PubMed]
- Van Montfrans, J.M.; Hoepelman, A.I.; Otto, S.; van Gijn, M.; van de Corput, L.; de Weger, R.A.; Monaco-Shawver, L.; Banerjee, P.P.; Sanders, E.A.; Jol-van der Zijde, C.M.; et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J. Allergy Clin. Immunol. 2012, 129, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Mayor, P.C.; Eng, K.H.; Singel, K.L.; Abrams, S.I.; Odunsi, K.; Moysich, K.B.; Fuleihan, R.; Garabedian, E.; Lugar, P.; Ochs, H.D.; et al. Cancer in primary immunodeficiency diseases: Cancer incidence in the United States Immune Deficiency Network Registry. J. Allergy Clin. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mellemkjaer, L.; Hammarstrom, L.; Andersen, V.; Yuen, J.; Heilmann, C.; Barington, T.; Bjorkander, J.; Olsen, J.H. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: A combined Danish and Swedish study. Clin. Exp. Immunol. 2002, 130, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, F.; da Silva, S.P.; Lucas, M.; Travis, S.; Chapel, H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin. Exp. Immunol. 2011, 165, 1–7. [Google Scholar] [CrossRef] [PubMed]
- IARC Helicobacter pylori Working Group. Helicobacter pylori Eradication as a Strategy for Gastric Cancer Prevention; IARC Working Group Reports, No. 8; International Agency for Research on Cancer: Lyon, France, 2014; Available online: http://www.iarc.fr/en/publications/pdfs-online/wrk/wrk8/index.php (acccessed on 17 September 2016).
- Danesh, J. Helicobacter pylori infection and gastric cancer: Systematic review of the epidemiological studies. Aliment. Pharmacol. Ther. 1999, 13, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001, 49, 347–353. [Google Scholar]
- Quiding-Jarbrink, M.; Sundstrom, P.; Lundgren, A.; Hansson, M.; Backstrom, M.; Johansson, C.; Enarsson, K.; Hermansson, M.; Johnsson, E.; Svennerholm, A.M. Decreased IgA antibody production in the stomach of gastric adenocarcinoma patients. Clin. Immunol. 2009, 131, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Lee, J.; Sano, T.; Janjigian, Y.Y.; Fan, D.; Song, S. Gastric adenocarcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17036. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Yu, X.J.; Zhan, S.H.; Jia, S.J.; Tian, Z.B.; Dong, Q.J. Participation of microbiota in the development of gastric cancer. World J. Gastroenterol. 2014, 20, 4948–4952. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Qu, Y.H.; Chu, X.D.; Wang, R.; Nelson, H.H.; Gao, Y.T.; Yuan, J.M. Urinary levels of N-nitroso compounds in relation to risk of gastric cancer: Findings from the shanghai cohort study. PLoS ONE 2015, 10, e0117326. [Google Scholar] [CrossRef] [PubMed]
- De Petris, G.; Dhungel, B.M.; Chen, L.; Chang, Y.H. Gastric adenocarcinoma in common variable immunodeficiency: Features of cancer and associated gastritis may be characteristic of the condition. Int. J. Surg. Pathol. 2014, 22, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Coati, I.; Fassan, M.; Farinati, F.; Graham, D.Y.; Genta, R.M.; Rugge, M. Autoimmune gastritis: Pathologist’s viewpoint. World J. Gastroenterol. 2015, 21, 12179–12189. [Google Scholar] [CrossRef] [PubMed]
- Kulnigg-Dabsch, S. Autoimmune gastritis. Wien. Med. Wochenschr. 2016, 166, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Bergman, M.P.; Faller, G.; D’Elios, M.M.; Del Prete, G.; Vandenbroucke-Grauls, C.M.J.E.; Appelmelk, B.J. Gastric automminity. In Helicobacter pylori: Physiology and Genetics; Mobley, H.L.T., Mendz, G.L., Hazell, S.L., Eds.; ASM Press: Washington, DC, USA, 2001; Chapter 36. [Google Scholar]
- Morimoto, Y.; Routes, J.M. Granulomatous disease in common variable immunodeficiency. Curr. Allergy Asthma Rep. 2005, 5, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.A.; Lederman, H.M.; Maitra, A.; Montgomery, E.A. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): A clinicopathologic study and review. Am. J. Surg. Pathol. 2007, 31, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Hamashima, C. Current issues and future perspectives of gastric cancer screening. World J. Gastroenterol. 2014, 20, 13767–13774. [Google Scholar] [CrossRef] [PubMed]
- Calvet, X.; Sanchez-Delgado, J.; Montserrat, A.; Lario, S.; Ramirez-Lazaro, M.J.; Quesada, M.; Casalots, A.; Suarez, D.; Campo, R.; Brullet, E.; et al. Accuracy of diagnostic tests for Helicobacter pylori: A reappraisal. Clin. Infect. Dis. 2009, 48, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, J.P.; Pajares, J.M. Stool antigen test for the diagnosis of Helicobacter pylori infection: A systematic review. Helicobacter 2004, 9, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Niv, Y. H pylori recurrence after successful eradication. World J. Gastroenterol. 2008, 14, 1477–1478. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chiang, T.H.; Liou, J.M.; Chen, H.H.; Wu, M.S.; Graham, D.Y. Mass Eradication of Helicobacter pylori to Prevent Gastric Cancer: Theoretical and Practical Considerations. Gut Liver 2016, 10, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Ribeiro, M.; Lopes, C.; da Costa-Pereira, A.; Guilherme, M.; Barbosa, J.; Lomba-Viana, H.; Silva, R.; Moreira-Dias, L. A follow up model for patients with atrophic chronic gastritis and intestinal metaplasia. J. Clin. Pathol. 2004, 57, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jeon, S.W.; Jung, M.K.; Cho, C.M.; Tak, W.Y.; Kweon, Y.O.; Kim, S.K.; Choi, Y.H. Long-term follow-up study of gastric intraepithelial neoplasias: Progression from low-grade dysplasia to invasive carcinoma. Eur. J. Gastroenterol. Hepatol. 2008, 20, 966–970. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.C.; Haringsma, J.; Kuipers, E.J. The detection, surveillance and treatment of premalignant gastric lesions related to Helicobacter pylori infection. Helicobacter 2007, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gompels, M.M.; Hodges, E.; Lock, R.J.; Angus, B.; White, H.; Larkin, A.; Chapel, H.M.; Spickett, G.P.; Misbah, S.A.; Smith, J.L.; et al. Lymphoproliferative disease in antibody deficiency: A multi-centre study. Clin. Exp. Immunol. 2003, 134, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Unger, S.; Seidl, M.; Schmitt-Graeff, A.; Bohm, J.; Schrenk, K.; Wehr, C.; Goldacker, S.; Drager, R.; Gartner, B.C.; Fisch, P.; et al. Ill-defined germinal centers and severely reduced plasma cells are histological hallmarks of lymphadenopathy in patients with common variable immunodeficiency. J. Clin. Immunol. 2014, 34, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Wehr, C.; Kivioja, T.; Schmitt, C.; Ferry, B.; Witte, T.; Eren, E.; Vlkova, M.; Hernandez, M.; Detkova, D.; Bos, P.R.; et al. The EUROclass trial: Defining subgroups in common variable immunodeficiency. Blood 2008, 111, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Desar, I.M.; Keuter, M.; Raemaekers, J.M.; Jansen, J.B.; van Krieken, J.H.; van der Meer, J.W. Extranodal marginal zone (MALT) lymphoma in common variable immunodeficiency. Neth. J. Med. 2006, 64, 136–140. [Google Scholar] [PubMed]
- Cunningham-Rundles, C.; Bodian, C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin. Immunol. 1999, 92, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Chapel, H.; Cunningham-Rundles, C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br. J. Haematol. 2009, 145, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Rundles, C.; Lieberman, P.; Hellman, G.; Chaganti, R.S. Non-Hodgkin lymphoma in common variable immunodeficiency. Am. J. Hematol. 1991, 37, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wotherspoon, A.C.; Ortiz-Hidalgo, C.; Falzon, M.R.; Isaacson, P.G. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991, 338, 1175–1176. [Google Scholar] [CrossRef]
- Gottesman, S.R.; Haas, D.; Ladanyi, M.; Amorosi, E.L. Peripheral T cell lymphoma in a patient with common variable immunodeficiency disease: Case report and literature review. Leuk. Lymphoma 1999, 32, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Jesus, A.A.; Jacob, C.M.; Silva, C.A.; Dorna, M.; Pastorino, A.C.; Carneiro-Sampaio, M. Common variable immunodeficiency associated with hepatosplenic T-cell lymphoma mimicking juvenile systemic lupus erythematosus. Clin. Dev. Immunol. 2011, 2011, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gammon, B.; Robson, A.; Deonizio, J.; Arkin, L.; Guitart, J. CD8+ granulomatous cutaneous T-cell lymphoma: A potential association with immunodeficiency. J. Am. Acad. Dermatol. 2014, 71, 555–560. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Defect | References |
|---|---|---|
| TNFRSF13B | Homozygous and heterozygous mutations | [18,19,24] |
| BAFF-R | Homozygous and heterozygous mutations | [25] |
| CD20 | Homozygous mutations | [29] |
| CD19-B-cell receptor complex | Homozygous mutations | [30,31] |
| ICOS | Homozygous deletions | [32] |
| Genes implicated in DNA repair (MSH5, MSH2, MLH1, RAD50 and NBS1) | Heterozygous non-synonymous mutations | [35] |
| CARD11 | Heterozygous single nucleotide polymorphisms | [36] |
| Bob1 | Heterozygous single nucleotide polymorphisms | [36] |
| MHC region | Single nucleotide polymorphisms | [37] |
| ADAM | Single nucleotide polymorphisms | [37] |
| CTLA4 | Heterozygous nonsense mutations Frameshift deletion Intronic mutations | [38] |
| PIK3CD | Heterozygous splice site mutations Gain-of-function mutations | [39] |
| NFκB2 | Heterozygous frameshift mutation Heterozygous nonsense mutation | [40] |
| PLCG2 | Deletions | [41] |
| LRBA | Homozygous mutations | [42] |
| CD27 | Homozygous mutations | [43] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leone, P.; Vacca, A.; Dammacco, F.; Racanelli, V. Common Variable Immunodeficiency and Gastric Malignancies. Int. J. Mol. Sci. 2018, 19, 451. https://doi.org/10.3390/ijms19020451
Leone P, Vacca A, Dammacco F, Racanelli V. Common Variable Immunodeficiency and Gastric Malignancies. International Journal of Molecular Sciences. 2018; 19(2):451. https://doi.org/10.3390/ijms19020451
Chicago/Turabian StyleLeone, Patrizia, Angelo Vacca, Franco Dammacco, and Vito Racanelli. 2018. "Common Variable Immunodeficiency and Gastric Malignancies" International Journal of Molecular Sciences 19, no. 2: 451. https://doi.org/10.3390/ijms19020451
APA StyleLeone, P., Vacca, A., Dammacco, F., & Racanelli, V. (2018). Common Variable Immunodeficiency and Gastric Malignancies. International Journal of Molecular Sciences, 19(2), 451. https://doi.org/10.3390/ijms19020451