Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease


Abstract
1. Introduction
2. Hypoxia-Inducible Factor
2.1. Regulation of HIF
2.2. Genetic Determinants of HIF Activity
2.3. HIF and Iron Handling
3. Current Standard of Care for Anemia in Chronic Kidney Disease (CKD)
3.1. Pitfalls of Current Standards of Care
3.2. Treatment to Hemoglobin Targets
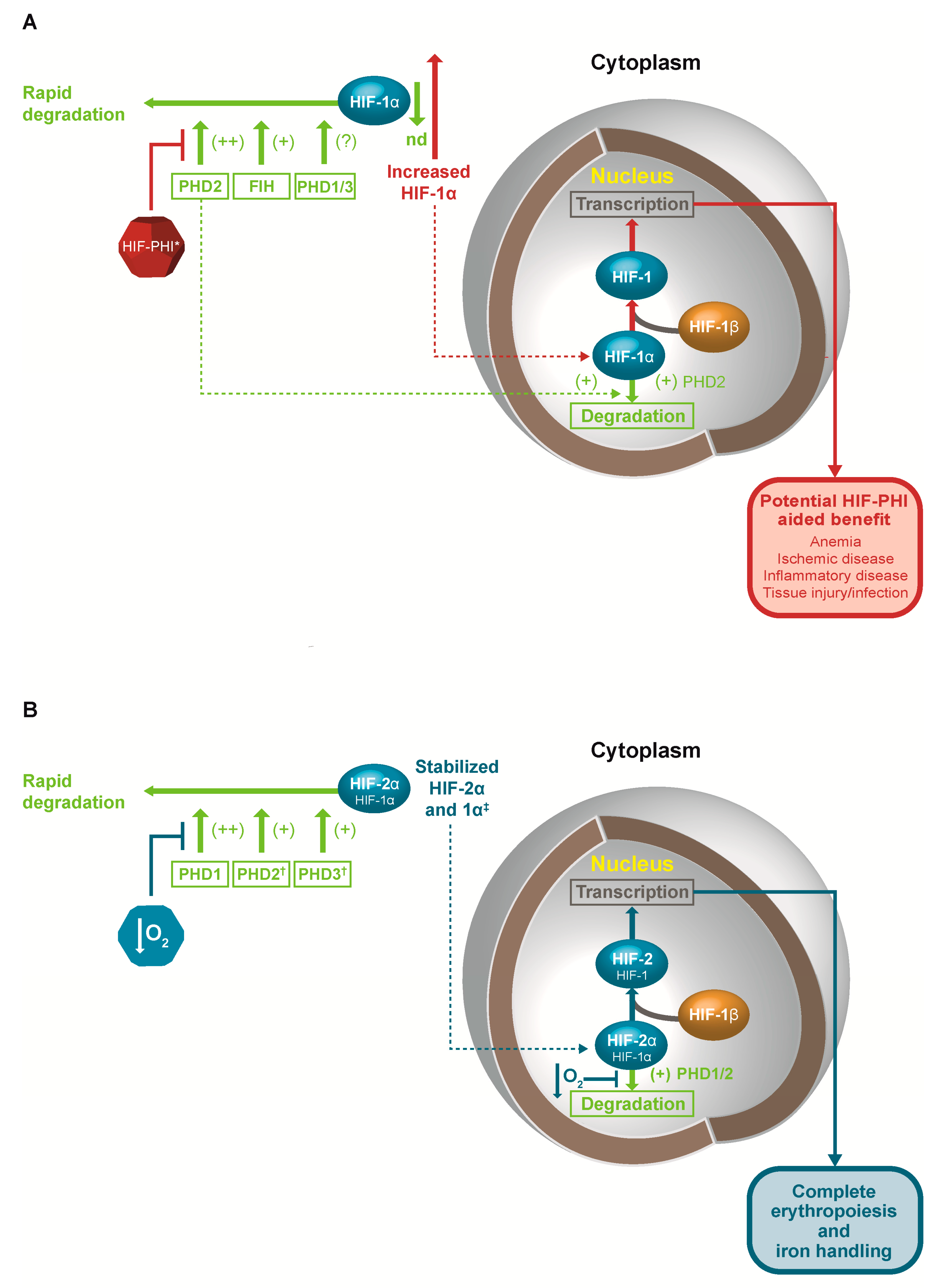
3.3. Hypoxia-Inducible Factor Prolyl-Hydroxylase Inhibitors
“New agents under development to pharmacologically manipulate HIF present exciting new possibilities in the treatment of anemia of chronic kidney disease.”
3.4. Clinical Impact of HIF-PHIs in Anemia of CKD
“Based on mode of action and available evidence, treatment with HIF-PHIs may allay concerns evident with current standards of care.”
4. Other Potential Therapeutic Roles for HIF-PHIs
“Clinical relevance remains speculative but new HIF-PHIs may provide benefits in a broad spectrum of disease states.”
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Locatelli, F.; Barany, P.; Covic, A.; de Francisco, A.; del Vecchio, L.; Goldsmith, D.; Horl, W.; London, G.; Vanholder, R.; van Biesen, W. Kidney disease: Improving global outcomes guidelines on anaemia management in chronic kidney disease: A European renal best practice position statement. Nephrol. Dial. Transplant 2013, 28, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E. Anaemia, diabetes and chronic kidney disease: Where are we now? J. Ren. Care 2012, 38, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Koulouridis, I.; Alfayez, M.; Trikalinos, T.A.; Balk, E.M.; Jaber, B.L. Dose of erythropoiesis-stimulating agents and adverse outcomes in CKD: A metaregression analysis. Am. J. Kidney Dis. 2013, 61, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Safety issues in iron treatment in CKD. Semin. Nephrol. 2016, 36, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wish, J.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Heidbreder, M.; Frohlich, F.; Johren, O.; Dendorfer, A.; Qadri, F.; Dominiak, P. Hypoxia rapidly activates HIF-3α mRNA expression. FASEB J. 2003, 17, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell. Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Drevytska, T.; Gavenauskas, B.; Drozdovska, S.; Nosar, V.; Dosenko, V.; Mankovska, I. HIF-3α mRNA expression changes in different tissues and their role in adaptation to intermittent hypoxia and physical exercise. Pathophysiology 2012, 19, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Soilleux, E.J.; Turley, H.; Tian, Y.M.; Pugh, C.W.; Gatter, K.C.; Harris, A.L. Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology 2005, 47, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, K.; Kowata, Y.; Yoshida, A.; Torii, S.; Sogawa, K. Role of the intracellular localization of HIF-prolyl hydroxylases. Biochim. Biophys. Acta 2009, 1793, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, M.E.; Fischer, H.; Poellinger, L.; Johnson, R.S.; Gustafsson, T.; Sundberg, C.J.; Rundqvist, H. Negative regulation of HIF in skeletal muscle of elite endurance athletes: A tentative mechanism promoting oxidative metabolism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R248–255. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.; Roux, D.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α. Cancer Res. 2006, 66, 3688–3698. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Ilott, N.E.; Schodel, J.; Sims, D.; Tumber, A.; Lippl, K.; Mole, D.R.; Pugh, C.W.; Ratcliffe, P.J.; Ponting, C.P.; et al. Tuning the transcriptional response to hypoxia by inhibiting hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases. J. Biol. Chem. 2016, 291, 20661–20673. [Google Scholar] [CrossRef] [PubMed]
- Ehrismann, D.; Flashman, E.; Genn, D.N.; Mathioudakis, N.; Hewitson, K.S.; Ratcliffe, P.J.; Schofield, C.J. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 2007, 401, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Page, E.L.; Chan, D.A.; Giaccia, A.J.; Levine, M.; Richard, D.E. Hypoxia-inducible factor-1α stabilization in nonhypoxic conditions: Role of oxidation and intracellular ascorbate depletion. Mol. Biol. Cell. 2008, 19, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Van Geyte, K.; Fraisl, P.; Kiss, J.; Aragones, J.; Mazzone, M.; Mairbaurl, H.; De Bock, K.; Jeoung, N.H.; Mollenhauer, M.; et al. Loss or silencing of the PHD1 prolyl hydroxylase protects livers of mice against ischemia/reperfusion injury. Gastroenterology 2010, 138, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Adluri, R.S.; Thirunavukkarasu, M.; Dunna, N.R.; Zhan, L.; Oriowo, B.; Takeda, K.; Sanchez, J.A.; Otani, H.; Maulik, G.; Fong, G.H.; et al. Disruption of hypoxia-inducible transcription factor-prolyl hydroxylase domain-1 (PHD-1−/−) attenuates ex vivo myocardial ischemia/reperfusion injury through hypoxia-inducible factor-1α transcription factor and its target genes in mice. Antioxid. Redox. Signal. 2011, 15, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Quaegebeur, A.; Segura, I.; Schmieder, R.; Verdegem, D.; Decimo, I.; Bifari, F.; Dresselaers, T.; Eelen, G.; Ghosh, D.; Davidson, S.M.; et al. Deletion or inhibition of the oxygen sensor PHD1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell. Metab. 2016, 23, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Belaidi, E.; Aron-Wisnewsky, J.; van der Zon, G.C.; Levy, P.; Clement, K.; Pepin, J.L.; Godin-Ribuot, D.; Guigas, B. Hypoxia-inducible factor prolyl hydroxylase 1 (PHD1) deficiency promotes hepatic steatosis and liver-specific insulin resistance in mice. Sci. Rep. 2016, 6, 24618. [Google Scholar] [CrossRef] [PubMed]
- Marsch, E.; Demandt, J.A.; Theelen, T.L.; Tullemans, B.M.; Wouters, K.; Boon, M.R.; van Dijk, T.H.; Gijbels, M.J.; Dubois, L.J.; Meex, S.J.; et al. Deficiency of the oxygen sensor prolyl hydroxylase 1 attenuates hypercholesterolaemia, atherosclerosis, and hyperglycaemia. Eur. Heart J. 2016, 37, 2993–2997. [Google Scholar] [CrossRef] [PubMed]
- Rishi, M.T.; Selvaraju, V.; Thirunavukkarasu, M.; Shaikh, I.A.; Takeda, K.; Fong, G.H.; Palesty, J.A.; Sanchez, J.A.; Maulik, N. Deletion of prolyl hydroxylase domain proteins (PHD1, PHD3) stabilizes hypoxia inducible factor-1α, promotes neovascularization, and improves perfusion in a murine model of hind-limb ischemia. Microvasc. Res. 2015, 97, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, M.; Kiss, J.; Dudda, J.; Kirchberg, J.; Rahbari, N.; Radhakrishnan, P.; Niemietz, T.; Rausch, V.; Weitz, J.; Schneider, M. Deficiency of the oxygen sensor PHD1 augments liver regeneration after partial hepatectomy. Langenbecks Arch. Surg. 2012, 397, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cowan, A.; Fong, G.H. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 2007, 116, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Aguila, H.L.; Parikh, N.S.; Li, X.; Lamothe, K.; Duan, L.J.; Takeda, H.; Lee, F.S.; Fong, G.H. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 2008, 111, 3229–3235. [Google Scholar] [CrossRef] [PubMed]
- Rauner, M.; Franke, K.; Murray, M.; Singh, R.P.; Hiram-Bab, S.; Platzbecker, U.; Gassmann, M.; Socolovsky, M.; Neumann, D.; Gabet, Y.; et al. Increased EPO levels are associated with bone loss in mice lacking PHD2 in EPO-producing cells. J. Bone Miner. Res. 2016, 31, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Xing, W.; Pourteymoor, S.; Schulte, J.; Mohan, S. Conditional deletion of prolyl hydroxylase domain-containing protein 2 (PHD2) gene reveals its essential role in chondrocyte function and endochondral bone formation. Endocrinology 2016, 157, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, P.R.; Pei, F.; Lee, R.; Kerestes, H.; Percy, M.J.; Keith, B.; Simon, M.C.; Lappin, T.R.; Khurana, T.S.; Lee, F.S. A knock-in mouse model of human PHD2 gene-associated erythrocytosis establishes a haploinsufficiency mechanism. J. Biol. Chem. 2013, 288, 33571–33584. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Kalucka, J.; Mamlouk, S.; Singh, R.P.; Muschter, A.; Weidemann, A.; Iyengar, V.; Jahn, S.; Wieczorek, K.; Geiger, K.; et al. HIF-1α is a protective factor in conditional PHD2-deficient mice suffering from severe HIF-2α-induced excessive erythropoiesis. Blood 2013, 121, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- Oriowo, B.; Thirunavukkarasu, M.; Selvaraju, V.; Adluri, R.S.; Zhan, L.; Takeda, K.; Fong, G.H.; Sanchez, J.A.; Maulik, N. Targeted gene deletion of prolyl hydroxylase domain protein 3 triggers angiogenesis and preserves cardiac function by stabilizing hypoxia inducible factor 1α following myocardial infarction. Curr. Pharm. Des. 2014, 20, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Pi, X.; Wang, Z.; He, J.; Willis, M.S.; Patterson, C. Depletion of PHD3 protects heart from ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis. J. Mol. Cell. Cardiol. 2015, 80, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Bishop, T.; Gallagher, D.; Pascual, A.; Lygate, C.A.; de Bono, J.P.; Nicholls, L.G.; Ortega-Saenz, P.; Oster, H.; Wijeyekoon, B.; Sutherland, A.I.; et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol. Cell. Biol. 2008, 28, 3386–3400. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through PHD3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 2013, 19, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.T.; Garvalov, B.K.; Seidel, S.; Cuesta, A.M.; Ritter, M.; Filatova, A.; Foss, F.; Dopeso, H.; Essmann, C.L.; Maxwell, P.H.; et al. Loss of PHD3 allows tumours to overcome hypoxic growth inhibition and sustain proliferation through eGFR. Nat. Commun. 2014, 5, 5582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Fu, Z.; Linke, S.; Chicher, J.; Gorman, J.J.; Visk, D.; Haddad, G.G.; Poellinger, L.; Peet, D.J.; Powell, F.; et al. The asparaginyl hydroxylase factor inhibiting HIF-1α is an essential regulator of metabolism. Cell. Metab. 2010, 11, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.J.; Zhao, Q.; Flores, A.; Harrison, C.; Lappin, T.R.; Maxwell, P.H.; McMullin, M.F.; Lee, F.S. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, R.; Sutherland, S.; van Wesel, A.C.; Huizinga, E.G.; Percy, M.J.; Bierings, M.; Lee, F.S. Erythrocytosis associated with a novel missense mutation in the HIF2A gene. Haematologica 2010, 95, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Miasnikova, G.Y.; Sergueeva, A.I.; Niu, X.; Nouraie, M.; Okhotin, D.J.; Polyakova, L.A.; Ammosova, T.; Nekhai, S.; Ganz, T.; et al. Chuvash polycythemia VHLR200W mutation is associated with down-regulation of hepcidin expression. Blood 2011, 118, 5278–5282. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Ding, K.; Zhang, Q.; Oktay, Y.; Bennett, M.J.; Bennett, M.; Shelton, J.M.; Richardson, J.A.; Moe, O.; Garcia, J.A. HIF-2α regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood 2005, 105, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.; Hu, C.J.; Johnson, R.S.; Brown, E.J.; Keith, B.; Simon, M.C. Acute postnatal ablation of HIF-2α results in anemia. Proc. Natl. Acad. Sci. USA 2007, 104, 2301–2306. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2α mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Teofili, L.; Cenci, T.; Giona, F.; Torti, L.; Rea, M.; Foa, R.; Leone, G.; Larocca, L.M. A novel heterozygous HIF2AM535I mutation reinforces the role of oxygen sensing pathway disturbances in the pathogenesis of familial erythrocytosis. Haematologica 2008, 93, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Furlow, P.W.; Percy, M.J.; Sutherland, S.; Bierl, C.; McMullin, M.F.; Master, S.R.; Lappin, T.R.; Lee, F.S. Erythrocytosis-associated HIF-2α mutations demonstrate a critical role for residues C-terminal to the hydroxylacceptor proline. J. Biol. Chem. 2009, 284, 9050–9058. [Google Scholar] [CrossRef] [PubMed]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.; Klaus, S.J.; Bachmann, S. Hypoxia-inducible factor-2α-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010, 77, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Seagroves, T.N.; Ryan, H.E.; Lu, H.; Wouters, B.G.; Knapp, M.; Thibault, P.; Laderoute, K.; Johnson, R.S. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell. Biol. 2001, 21, 3436–3444. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell. Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Marce, M.; Okuyama, H.; Wesley, J.B.; Sarkar, K.; Kimura, H.; Liu, Y.V.; Zhang, H.; Strazza, M.; Rey, S.; Savino, L.; et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ. Res. 2007, 101, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.P.; Tournaire, R.; Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell. Physiol. 2008, 217, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Lieb, M.E.; Menzies, K.; Moschella, M.C.; Ni, R.; Taubman, M.B. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem. Cell. Biol. 2002, 80, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Tojo, Y.; Sekine, H.; Hirano, I.; Pan, X.; Souma, T.; Tsujita, T.; Kawaguchi, S.; Takeda, N.; Takeda, K.; Fong, G.H.; et al. Hypoxia signaling cascade for erythropoietin production in hepatocytes. Mol. Cell. Biol. 2015, 35, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, W.; Liu, X.; Zhang, Y. Tankyrase 1 polymorphism associated with an increased risk in developing non-small cell lung cancer in a Chinese population: A proof-of-principle study. Int. J. Clin. Exp. Pathol. 2015, 8, 10500–10511. [Google Scholar] [PubMed]
- Coleman, M.L.; McDonough, M.A.; Hewitson, K.S.; Coles, C.; Mecinovic, J.; Edelmann, M.; Cook, K.M.; Cockman, M.E.; Lancaster, D.E.; Kessler, B.M.; et al. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J. Biol. Chem. 2007, 282, 24027–24038. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Linke, S.; Dias, J.M.; Zheng, X.; Gradin, K.; Wallis, T.P.; Hamilton, B.R.; Gustafsson, M.; Ruas, J.L.; Wilkins, S.; et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 3368–3373. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Webb, J.D.; Kramer, H.B.; Kessler, B.M.; Ratcliffe, P.J. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteom. 2009, 8, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital disorder of oxygen sensing: Association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Muchnik, E.; Kaplan, J. HIF prolyl hydroxylase inhibitors for anemia. Expert Opin. Investig. Drugs 2011, 20, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.N.; Ponka, P. Identification of a hypoxia response element in the transferrin receptor gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Kvietikova, I.; Gassmann, M.; Wenger, R.H. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J. Biol. Chem. 1997, 272, 20055–20062. [Google Scholar] [CrossRef] [PubMed]
- Tacchini, L.; Bianchi, L.; Bernelli-Zazzera, A.; Cairo, G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J. Biol. Chem. 1999, 274, 24142–24146. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, C.K.; Mazumder, B.; Fox, P.L. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J. Biol. Chem. 2000, 275, 21048–21054. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; Grondin, F.; McDonald, P.P.; Richard, D.E.; Dubois, C.M. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: Impact on the bioactivation of proproteins. J. Biol. Chem. 2005, 280, 6561–6569. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Camaschella, C. Furin-mediated release of soluble hemojuvelin: A new link between hypoxia and iron homeostasis. Blood 2008, 111, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Hayat, A.; Haria, D.; Salifu, M.O. Erythropoietin stimulating agents in the management of anemia of chronic kidney disease. Patient Prefer. Adherence 2008, 2, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Besarab, A.; Ayyoub, F. Anemia in renal disease. In Diseases of the Kidney and Urinary Tract, 8th ed.; Schrier, R., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 2406–2430. [Google Scholar]
- Babitt, J.L.; Lin, H.Y. Molecular mechanisms of hepcidin regulation: Implications for the anemia of CKD. Am. J. Kidney Dis. 2010, 55, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Garcia-Erce, J.A.; Remacha, A.F. Disorders of iron metabolism. Part 1: Molecular basis of iron homoeostasis. J. Clin. Pathol. 2011, 64, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, V.S.; Ganji, S.H.; Shelkovnikov, S.; Norris, K.; Vaziri, N.D. Iron sucrose promotes endothelial injury and dysfunction and monocyte adhesion/infiltration. Am. J. Nephrol. 2012, 35, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Bistrian, B.R.; Khaodhiar, L. The systemic inflammatory response and its impact on iron nutriture in end-stage renal disease. Am. J. Kidney Dis. 1999, 34, S35–S39. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Oxidative stress in uremia: Nature, mechanisms, and potential consequences. Semin. Nephrol. 2004, 24, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P. Inflammation in end-stage renal disease: The hidden enemy. Nephrology 2006, 11, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, L.M.; Cardozo, L.F.; Medeiros, R.F.; Stockler-Pinto, M.B.; Mafra, D. Association between serum ferritin and lipid peroxidation in hemodialysis patients. J. Bras. Nefrol. 2015, 37, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Minqin, R.; Watt, F.; Huat, B.T.; Halliwell, B. Correlation of iron and zinc levels with lesion depth in newly formed atherosclerotic lesions. Free Radic. Biol. Med. 2003, 34, 746–752. [Google Scholar] [CrossRef]
- Murillo-Ortiz, B.; Ramirez Emiliano, J.; Hernandez Vazquez, W.I.; Martinez-Garza, S.; Solorio-Meza, S.; Albarran-Tamayo, F.; Ramos-Rodriguez, E.; Benitez-Bribiesca, L. Impact of oxidative stress in premature aging and iron overload in hemodialysis patients. Oxid. Med. Cell. Longev. 2016, 2016, 1578235. [Google Scholar] [CrossRef] [PubMed]
- Karaboyas, A.; Zee, J.; Morgenstern, H.; Nolen, J.G.; Hakim, R.; Kalantar-Zadeh, K.; Zager, P.; Pisoni, R.L.; Port, F.K.; Robinson, B.M. Understanding the recent increase in ferritin levels in United States dialysis patients: Potential impact of changes in intravenous iron and erythropoiesis-stimulating agent dosing. Clin. J. Am. Soc. Nephrol. 2015, 10, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.L.; Hung, S.C.; Lin, Y.P.; Tang, C.F.; Lee, T.S.; Lin, C.P.; Tarng, D.C. Intravenous ferric chloride hexahydrate supplementation induced endothelial dysfunction and increased cardiovascular risk among hemodialysis patients. PLoS ONE 2012, 7, e50295. [Google Scholar] [CrossRef] [PubMed]
- Kuragano, T.; Matsumura, O.; Matsuda, A.; Hara, T.; Kiyomoto, H.; Murata, T.; Kitamura, K.; Fujimoto, S.; Hase, H.; Joki, N.; et al. Association between hemoglobin variability, serum ferritin levels, and adverse events/mortality in maintenance hemodialysis patients. Kidney Int. 2014, 86, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Bailie, G.R.; Larkina, M.; Goodkin, D.A.; Li, Y.; Pisoni, R.L.; Bieber, B.; Mason, N.; Tong, L.; Locatelli, F.; Marshall, M.R.; et al. Data from the Dialysis Outcomes and Practice Patterns Study validate an association between high intravenous iron doses and mortality. Kidney Int. 2015, 87, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.; Witko-Sarsat, V.; Massy, Z.; Descamps-Latscha, B.; Guerin, A.P.; Marchais, S.J.; Gausson, V.; London, G.M. Iron therapy, advanced oxidation protein products, and carotid artery intima-media thickness in end-stage renal disease. Circulation 2002, 106, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Reis, K.A.; Guz, G.; Ozdemir, H.; Erten, Y.; Atalay, V.; Bicik, Z.; Ozkurt, Z.N.; Bali, M.; Sindel, S. Intravenous iron therapy as a possible risk factor for atherosclerosis in end-stage renal disease. Int. Heart J. 2005, 46, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Belo, L.; Reis, F.; Santos-Silva, A. Iron therapy in chronic kidney disease: Recent changes, benefits and risks. Blood Rev. 2016, 30, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Deicher, R.; Ziai, F.; Cohen, G.; Mullner, M.; Horl, W.H. High-dose parenteral iron sucrose depresses neutrophil intracellular killing capacity. Kidney Int. 2003, 64, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Porto, G.; de Sousa, M. Iron overload and immunity. World J. Gastroenterol. 2007, 13, 4707–4715. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Fonseca, V.A.; Alam, M.G.; Shah, S.V. The role of iron in diabetes and its complications. Diabetes Care 2007, 30, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Tang, X.; Fu, P.; Liu, F. Secondary haemochromatosis in a haemodialysis patient. Singap. Med. J. 2015, 56, e124–e126. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, C.; Inaba, M.; Ishimura, E.; Yamakawa, T.; Shoji, S.; Okuno, S. Association of increased serum ferritin with impaired muscle strength/quality in hemodialysis patients. J. Ren. Nutr. 2016, 26, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Rostoker, G.; Cohen, Y. Magnetic resonance imaging repercussions of intravenous iron products used for iron-deficiency anemia and dialysis-associated anemia. J. Comput. Assist. Tomogr. 2014, 38, 843–844. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C. Epoetin-induced pure red cell aplasia: Diagnosis and treatment. Curr. Opin. Nephrol. Hypertens. 2007, 16, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, M.E.; Fan, T. Prevalence of anemia in chronic kidney disease in the United States. PLoS ONE 2014, 9, e84943. [Google Scholar] [CrossRef] [PubMed]
- Eschbach, J.W.; Egrie, J.C.; Downing, M.R.; Browne, J.K.; Adamson, J.W. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. N. Engl. J. Med. 1987, 316, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, J.; Racine, N.; Barre, P.; Sniderman, A.D. Regression of left ventricular hypertrophy in dialysis patients following correction of anemia with recombinant human erythropoietin. Can. J. Cardiol. 1990, 6, 1–4. [Google Scholar] [PubMed]
- Farag, Y.M.; Keithi-Reddy, S.R.; Mittal, B.V.; Surana, S.P.; Addabbo, F.; Goligorsky, M.S.; Singh, A.K. Anemia, inflammation and health-related quality of life in chronic kidney disease patients. Clin. Nephrol. 2011, 75, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Gibertoni, D.; Mandreoli, M.; Rucci, P.; Fantini, M.P.; Rigotti, A.; Scarpioni, R.; Santoro, A. Excess mortality attributable to chronic kidney disease. Results from the PIRP project. J. Nephrol. 2016, 29, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Nseir, W.; Artul, S.; Nasrallah, N.; Mograbi, J.; Mahamid, M. Hospitalization and 1-year all-cause mortality in type 2 diabetic patients with chronic kidney disease at Stages 1 and 2: Effect of mild anemia. J. Diabetes 2016, 8, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, T.; Gejyo, F.; Nishi, S.; Iino, Y.; Watanabe, Y.; Suzuki, M.; Saito, A.; Akiba, T.; Hirakata, H.; Fukuhara, S.; et al. Positive outcomes of high hemoglobin target in patients with chronic kidney disease not on dialysis: A randomized controlled study. Ther. Apher. Dial. 2011, 15, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.F.; Pfeffer, M.A.; Feng, A.; Uno, H.; McMurray, J.J.; Toto, R.; Gandra, S.R.; Solomon, S.D.; Moustafa, M.; Macdougall, I.C.; et al. Darbepoetin alfa impact on health status in diabetes patients with kidney disease: A randomized trial. Clin. J. Am. Soc. Nephrol. 2011, 6, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.D.; Jassal, S.V.; Woodward, M.C.; Soroka, S.; McMahon, L.P. A randomised single-blind study to improve health-related quality of life by treating anaemia of chronic kidney disease with Aranesp(R) (darbepoetin alfa) in older people: STIMULATE. Int. Urol. Nephrol. 2014, 46, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Revicki, D.A.; Brown, R.E.; Feeny, D.H.; Henry, D.; Teehan, B.P.; Rudnick, M.R.; Benz, R.L. Health-related quality of life associated with recombinant human erythropoietin therapy for predialysis chronic renal disease patients. Am. J. Kidney Dis. 1995, 25, 548–554. [Google Scholar] [CrossRef]
- Benz, R.L.; Pressman, M.R.; Hovick, E.T.; Peterson, D.D. A preliminary study of the effects of correction of anemia with recombinant human erythropoietin therapy on sleep, sleep disorders, and daytime sleepiness in hemodialysis patients (The SLEEPO study). Am. J. Kidney Dis. 1999, 34, 1089–1095. [Google Scholar] [CrossRef]
- Johansen, K.L.; Finkelstein, F.O.; Revicki, D.A.; Evans, C.; Wan, S.; Gitlin, M.; Agodoa, I.L. Systematic review of the impact of erythropoiesis-stimulating agents on fatigue in dialysis patients. Nephrol. Dial. Transplant. 2012, 27, 2418–2425. [Google Scholar] [CrossRef] [PubMed]
- Besarab, A.; Bolton, W.K.; Browne, J.K.; Egrie, J.C.; Nissenson, A.R.; Okamoto, D.M.; Schwab, S.J.; Goodkin, D.A. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N. Engl. J. Med. 1998, 339, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Correction of anemia with epoetin alfa in chronic kidney disease. N. Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Burdmann, E.A.; Chen, C.Y.; Cooper, M.E.; de Zeeuw, D.; Eckardt, K.U.; Feyzi, J.M.; Ivanovich, P.; Kewalramani, R.; Levey, A.S.; et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N. Engl. J. Med. 2009, 361, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.B.; Locatelli, F.; Clyne, N.; Eckardt, K.U.; Macdougall, I.C.; Tsakiris, D.; Burger, H.U.; Scherhag, A. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N. Engl. J. Med. 2006, 355, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Uno, H.; Lewis, E.F.; Eckardt, K.U.; Lin, J.; Burdmann, E.A.; de Zeeuw, D.; Ivanovich, P.; Levey, A.S.; Parfrey, P.; et al. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N. Engl. J. Med. 2010, 363, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Brines, M. The therapeutic potential of erythropoiesis-stimulating agents for tissue protection: A tale of two receptors. Blood Purif. 2010, 29, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Brookhart, M.A.; Schneeweiss, S.; Avorn, J.; Bradbury, B.D.; Rothman, K.J.; Fischer, M.; Mehta, J.; Winkelmayer, W.C. The effect of altitude on dosing and response to erythropoietin in ESRD. J. Am. Soc. Nephrol. 2008, 19, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Winkelmayer, W.C.; Liu, J.; Brookhart, M.A. Altitude and all-cause mortality in incident dialysis patients. JAMA 2009, 301, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Besarab, A.; Provenzano, R.; Hertel, J.; Zabaneh, R.; Klaus, S.J.; Lee, T.; Leong, R.; Hemmerich, S.; Yu, K.H.; Neff, T.B. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol. Dial. Transplant. 2015, 30, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, R.; Besarab, A.; Wright, S.; Dua, S.; Zeig, S.; Nguyen, P.; Poole, L.; Saikali, K.G.; Saha, G.; Hemmerich, S.; et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: A phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. Am. J. Kidney Dis. 2016, 67, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Holdstock, L.; Meadowcroft, A.M.; Maier, R.; Johnson, B.M.; Jones, D.; Rastogi, A.; Zeig, S.; Lepore, J.J.; Cobitz, A.R. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J. Am. Soc. Nephrol. 2016, 27, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Brigandi, R.A.; Johnson, B.; Oei, C.; Westerman, M.; Olbina, G.; de Zoysa, J.; Roger, S.D.; Sahay, M.; Cross, N.; McMahon, L.; et al. A novel hypoxia-inducible factor-prolyl hydroxylase inhibitor (GSK1278863) for anemia in CKD: A 28-Day, phase 2A randomized trial. Am. J. Kidney Dis. 2016, 67, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, R.; Besarab, A.; Sun, C.H.; Diamond, S.A.; Durham, J.H.; Cangiano, J.L.; Aiello, J.R.; Novak, J.E.; Lee, T.; Leong, R.; et al. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor Roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Spinowitz, B.S.; Hartman, C.S.; Maroni, B.J.; Haase, V.H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Petruliene, K.; Ziginskiene, E.; Kuzminskis, V.; Nedzelskiene, I.; Bumblyte, I.A. Hepcidin serum levels and resistance to recombinant human erythropoietin therapy in hemodialysis patients. Medicina 2017, 53, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Besarab, A.; Chernyavskaya, E.; Motylev, I.; Shutov, E.; Kumbar, L.M.; Gurevich, K.; Chan, D.T.; Leong, R.; Poole, L.; Zhong, M.; et al. Roxadustat (FG-4592): Correction of anemia in incident dialysis patients. J. Am. Soc. Nephrol. 2016, 27, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yang, E.G. Recent advances in developing inhibitors for hypoxia-inducible factor prolyl hydroxylases and their therapeutic implications. Molecules 2015, 20, 20551–20568. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, Y.; Rychkov, G.Y.; Peet, D.J. Modulation of TRP channel activity by hydroxylation and its therapeutic potential. Pharmaceuticals 2017, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Keely, S.; Karhausen, J.; Gerich, M.E.; Furuta, G.T.; Colgan, S.P. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 2008, 134, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Seeballuck, F.; Keely, S.J.; Mangan, N.E.; Callanan, J.J.; Fallon, P.G.; Taylor, C.T. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008, 134, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Marks, E.; Goggins, B.J.; Cardona, J.; Cole, S.; Minahan, K.; Mateer, S.; Walker, M.M.; Shalwitz, R.; Keely, S. Oral delivery of prolyl hydroxylase inhibitor: AKB-4924 promotes localized mucosal healing in a mouse model of colitis. Inflamm. Bowel Dis. 2015, 21, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Cavadas, M.A.; Tambuwala, M.M.; Hams, E.; Rodriguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Grenz, A.; Gorzolla, I.C.; Schittenhelm, J.; Dalton, J.H.; Eltzschig, H.K. Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J. Immunol. 2011, 186, 4367–4374. [Google Scholar] [CrossRef] [PubMed]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Tambuwala, M.M.; Cummins, E.P.; Lenihan, C.R.; Kiss, J.; Stauch, M.; Scholz, C.C.; Fraisl, P.; Lasitschka, F.; Mollenhauer, M.; Saunders, S.P.; et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 2010, 139, 2093–2101. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Mouse Models | ||
|---|---|---|
| Gene | Outcome | Publication |
| PHD1 | Knockout (KO) protects from I/R injury in the liver and heart, reducing size of infarction and increasing scavenging of oxygen radicals, and protects against ischemic stroke | Schneider, M.; et al. Gastroenterology 2010 [28] |
| Adluri, R.S.; et al. Antioxid. Redox Signal 2011 [29] | ||
| Quaegebeur, A.; et al. Cell Metab. 2016 [30] | ||
| Metabolic disturbance; KO promotes liver steatosis and insulin resistance, with increased glycolysis; attenuated hypercholesterolemia and hyperglycemia | Thomas, A.; et al. Sci. Rep. 2016 [31] | |
| Marsch, E.; et al. Eur. Heart J. 2016 [32] | ||
| KO increases capillary and arteriolar density in response to ischemia | Rishi, M.T.; et al. Microvasc. Res. 2015 [33] | |
| KO increases hepatocyte proliferation and liver regeneration | Mollenhauer, M.; et al. Langenbeck’s Arch. Surg. 2012 [34] | |
| PHD2 | Conditional knockout (CKO) leads to increased angiogenesis and angiectasia | Takeda, K.; et al. Circulation 2007 [35] |
| CKO increases EPO levels and erythropoiesis | Takeda, K.; et al.; Blood 2008 [36] | |
| CKO in EPO-producing cells leads to decreased bone density, while CKO in chondrocytes leads to increased bone density | Rauner, M.; et al. J. Bone Miner. Res. 2016 [37] | |
| Cheng, S.; et al. Endocrinology 2016 [38] | ||
| PHD2 erythrocytosis | Arsenault, P.R.; et al. J. Biol. Chem. 2013 [39] | |
| Franke, K.; et al. Blood 2013 [40] | ||
| PHD3 | KO leads to increased angiogenesis, with increased cardiac function and decreased fibrosis after ischemic injury | Oriowo, B.; et al. Curr. Pharm. Des. 2014 [41] |
| Xie, L.; et al. J. Mol. Cell. Cardiol. 2015 [42] | ||
| Regulation of neuronal apoptosis; dysregulation of sympathoadrenal development | Bishop, T.; et al. Mol. Cell. Biol. 2008 [43] | |
| KO leads to decreased neuronal apoptosis but decreased sympathoadrenal function | Taniguchi, C.M.; et al. Nat. Med. 2013 [44] | |
| Knockdown in glioblastoma cells and KO in astrocytoma cells; increased tumor growth | Henze, A.T.; et al. Nat. Commun. 2014 [45] | |
| FIH-1 | KO causes decreased weight, increased metabolic rate, resistance to hepatic steatosis, and high fat diet-induced weight gain (occurs also with KO in neuronal cells) | Zhang, N.; et al. Cell Metab. 2010 [46] |
| HIF2A | HIF2A erythrocytosis | Tan, Q.; et al. J. Biol. Chem. 2013 [47] |
| Human Mutations | ||
| PHD2 | Mutation causing decreased function causes congenital erythrocytosis | Percy, M.J.; et al. Proc. Natl. Acad. Sci. USA 2006 [48] |
| HIF2A | Mutation decreasing binding to PHD2 and VHL causes erythrocytosis | Van Wijk, R.; et al. Haematologica 2010 [49] |
| Percy, M.J.; et al. NEJM 2008 [50] | ||
| VHL | Germline loss-of-function in VHL leading to erythrocytosis | Gordeuk; et al. Blood 2011 [51] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplan, J.M.; Sharma, N.; Dikdan, S. Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 389. https://doi.org/10.3390/ijms19020389
Kaplan JM, Sharma N, Dikdan S. Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease. International Journal of Molecular Sciences. 2018; 19(2):389. https://doi.org/10.3390/ijms19020389
Chicago/Turabian StyleKaplan, Joshua M., Neeraj Sharma, and Sean Dikdan. 2018. "Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease" International Journal of Molecular Sciences 19, no. 2: 389. https://doi.org/10.3390/ijms19020389
APA StyleKaplan, J. M., Sharma, N., & Dikdan, S. (2018). Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease. International Journal of Molecular Sciences, 19(2), 389. https://doi.org/10.3390/ijms19020389