Structural Studies of Predicted Ligand Binding Sites and Molecular Docking Analysis of Slc2a4 as a Therapeutic Target for the Treatment of Cancer

 ,
,  ,
,
Abstract
:
1. Introduction
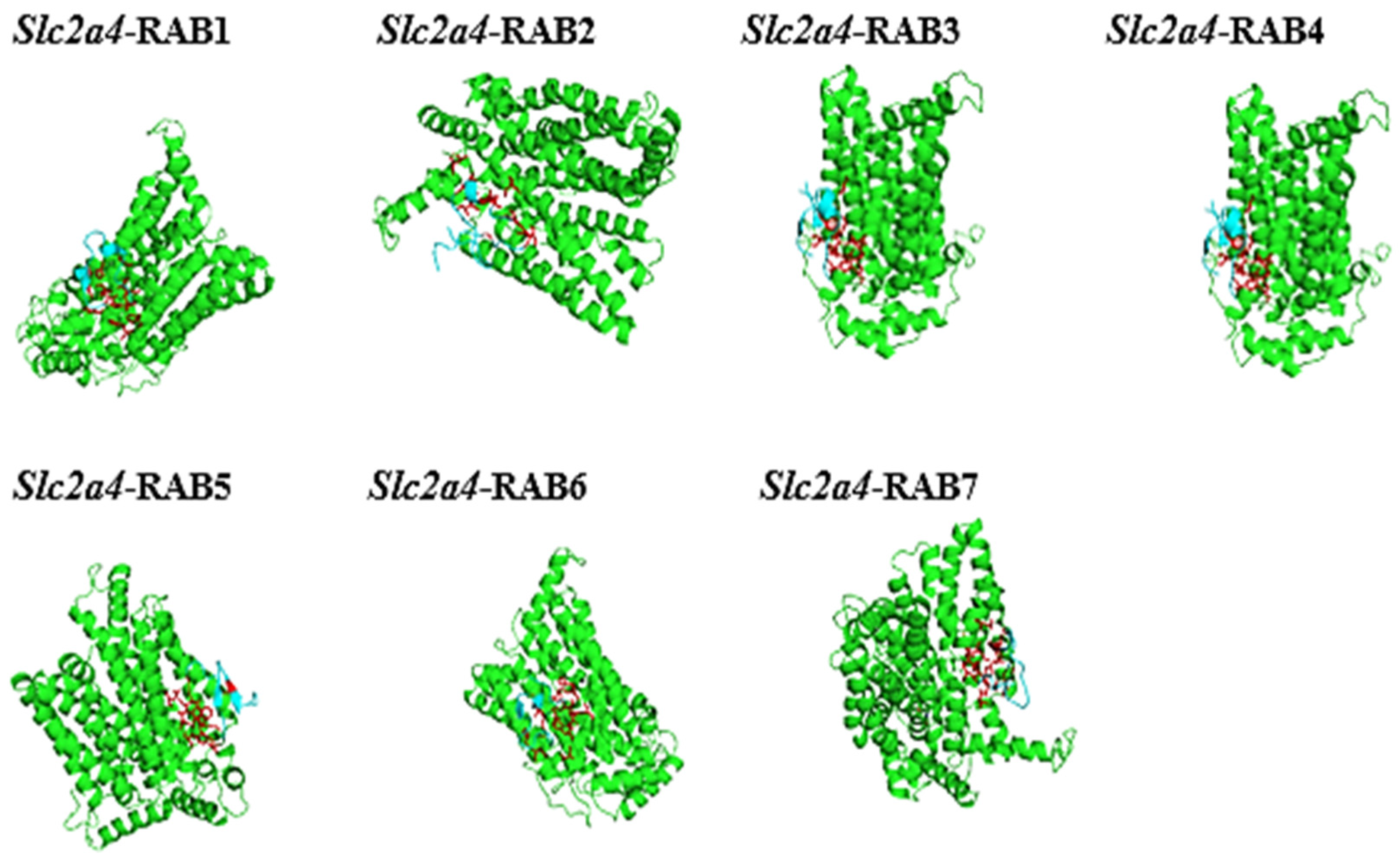
2. Results
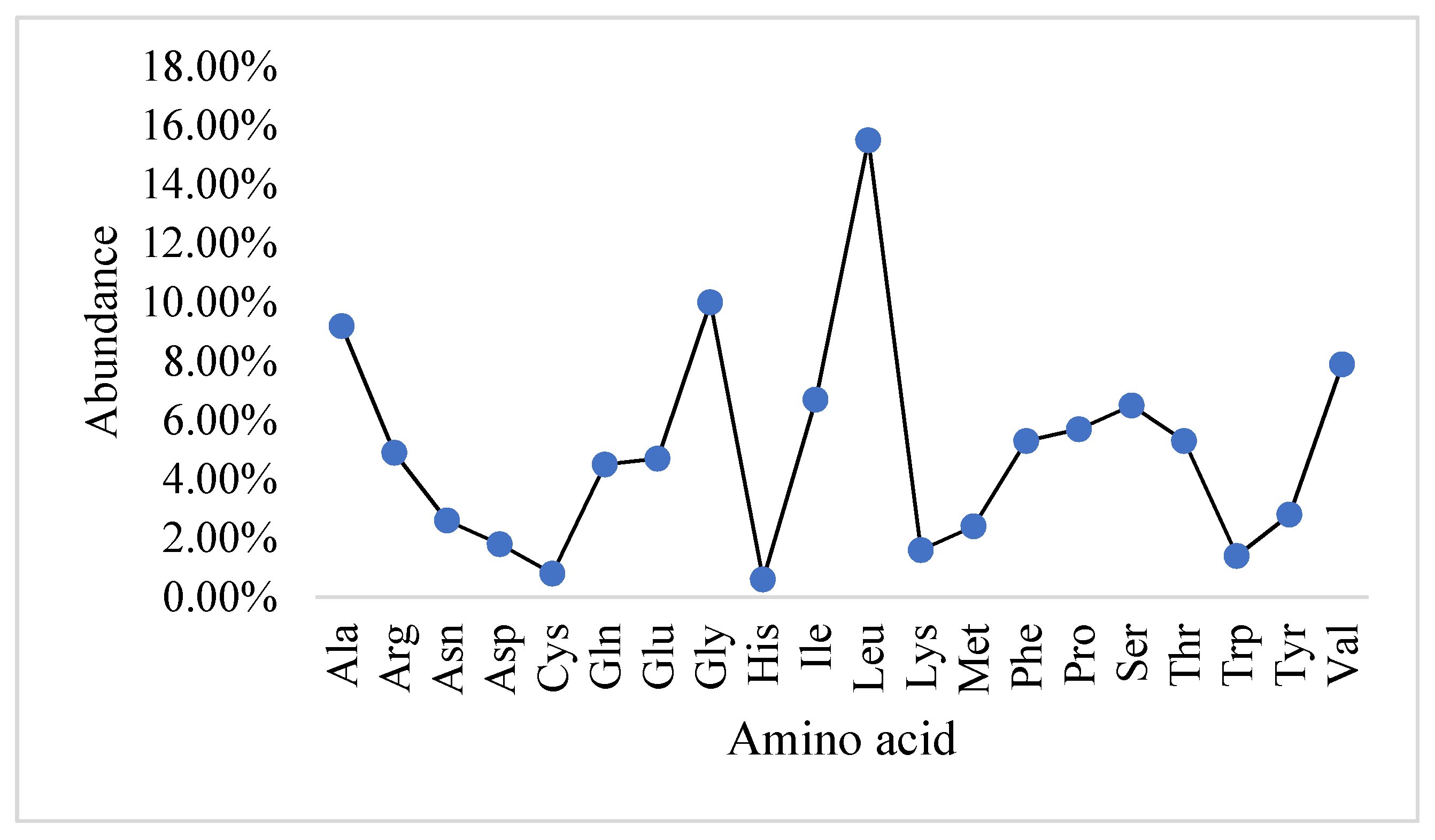
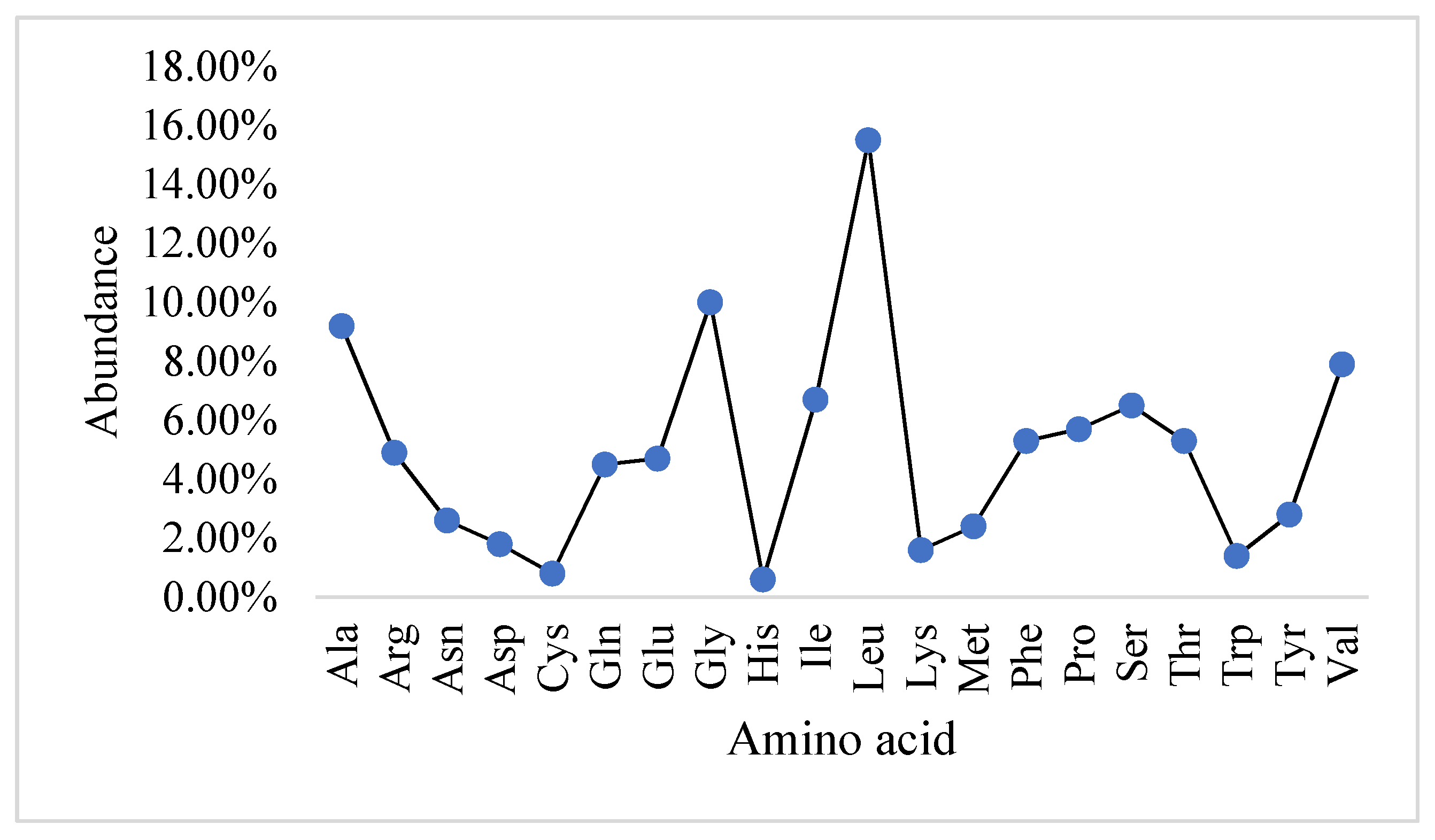
2.1. Physicochemical Properties and the Abundance of Amino Acids in Slc2a4 Protein
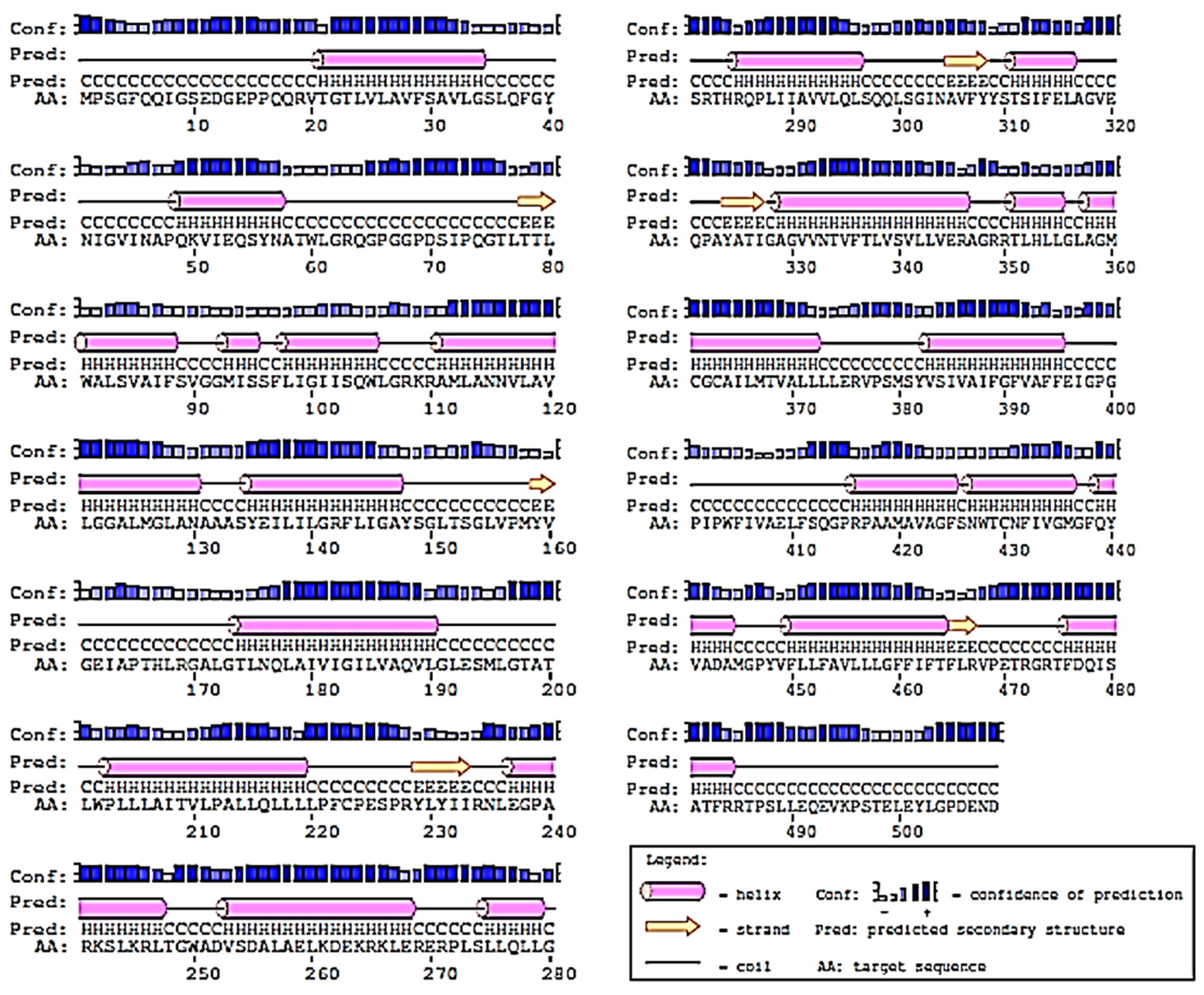
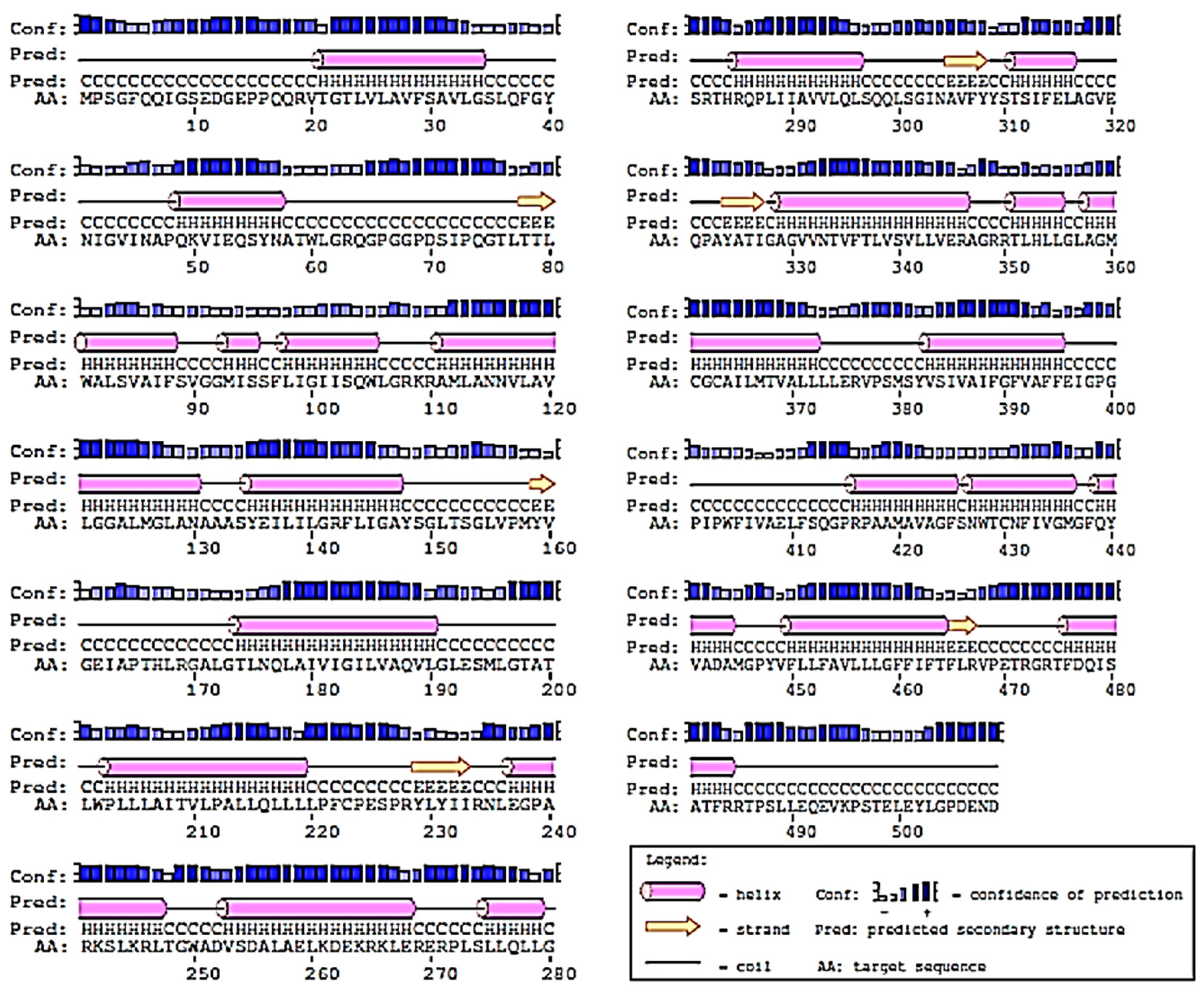
2.2. Prediction of 2D-Structure of Slc2a4 Protein
2.3. Discovery of Putative AMPs and Physicochemical Characterization
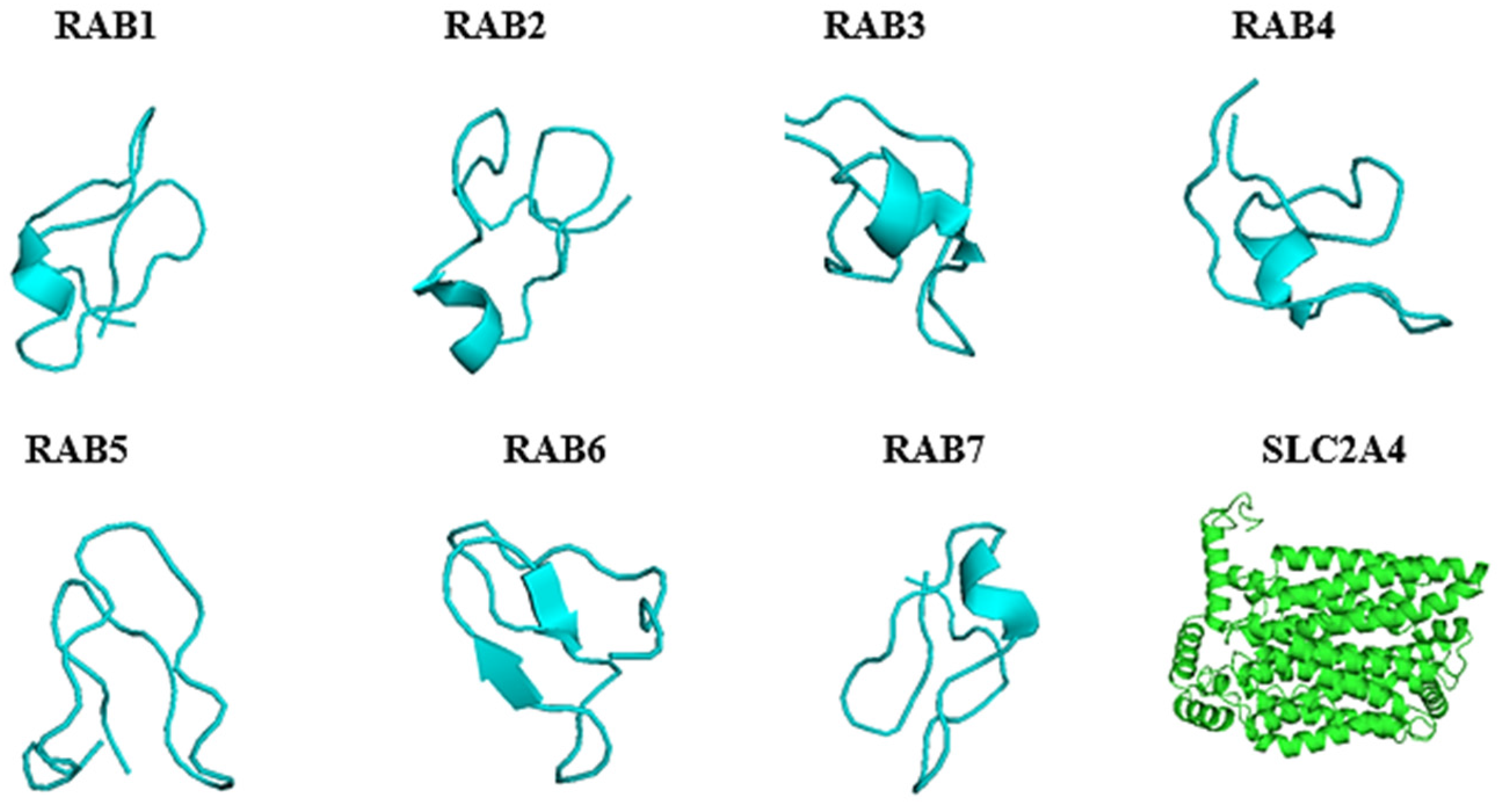
2.4. Predicted 3D Homology Model of the Anti-Cancer Putative AMPs and Slc2a4 by I-TASSER
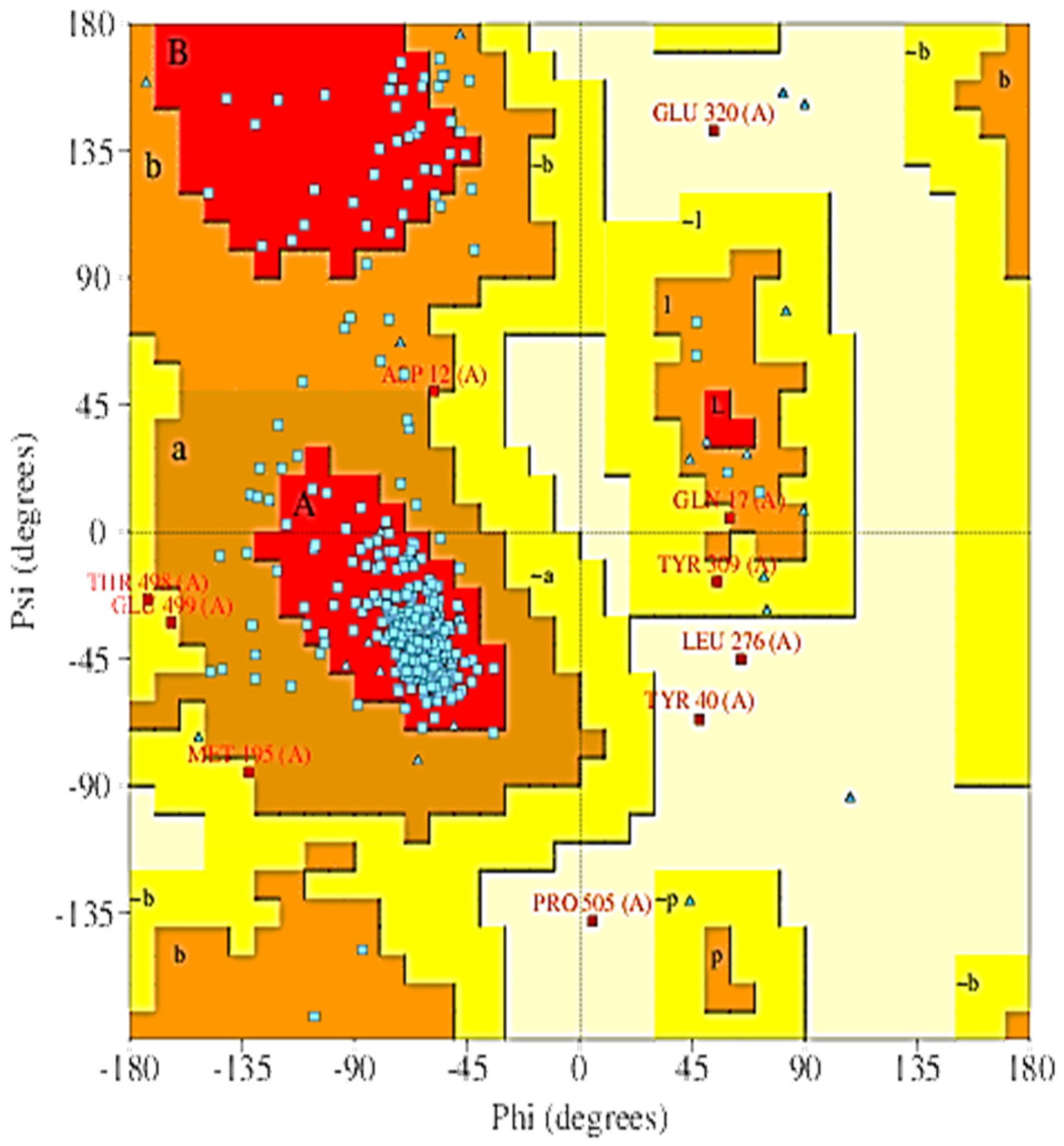
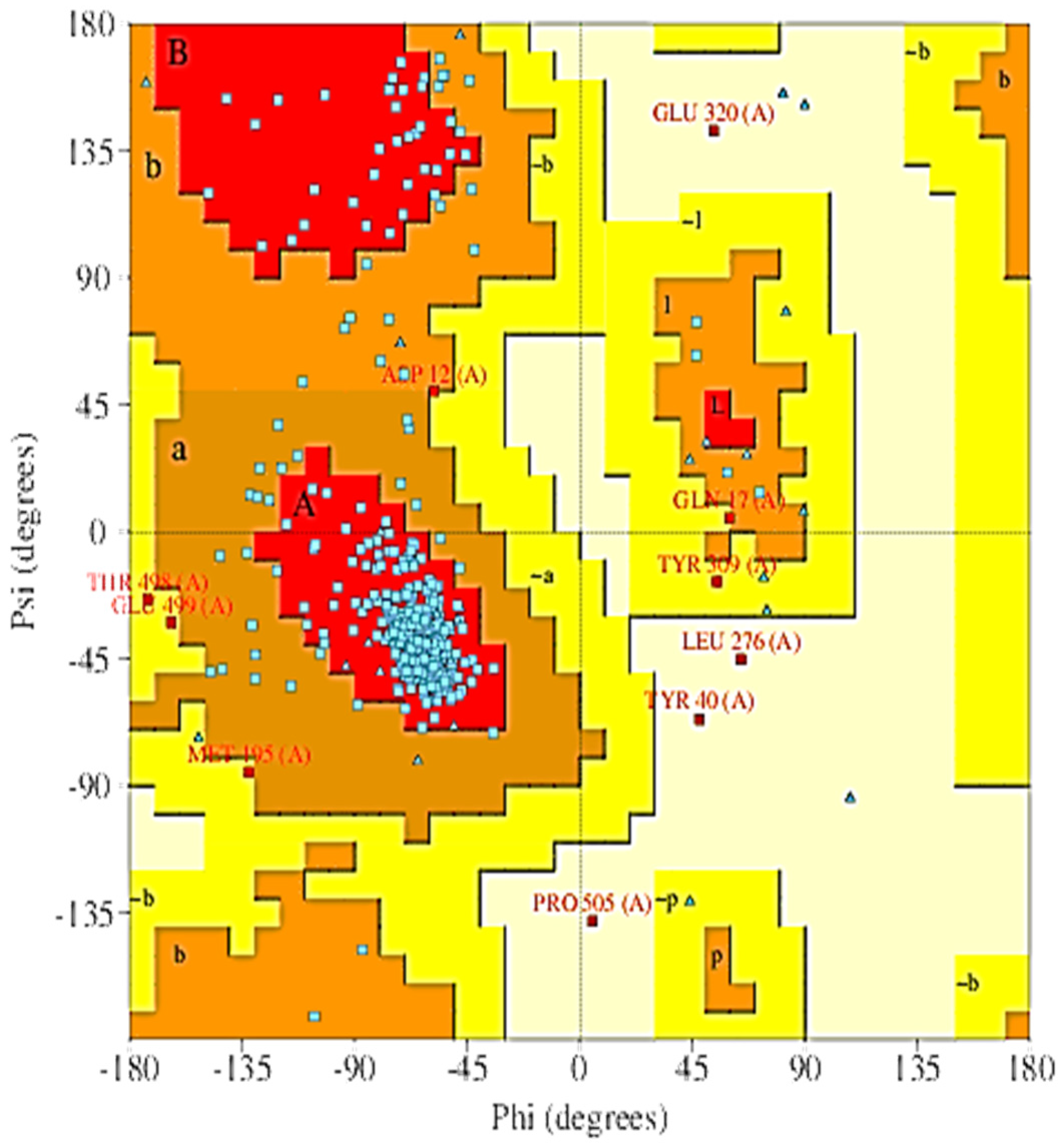
2.5. The 3D Modelled Structure Validation
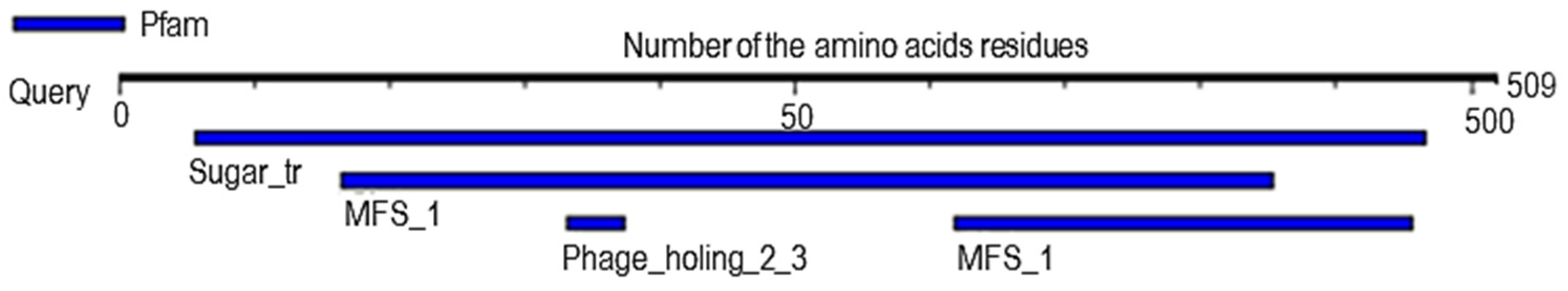
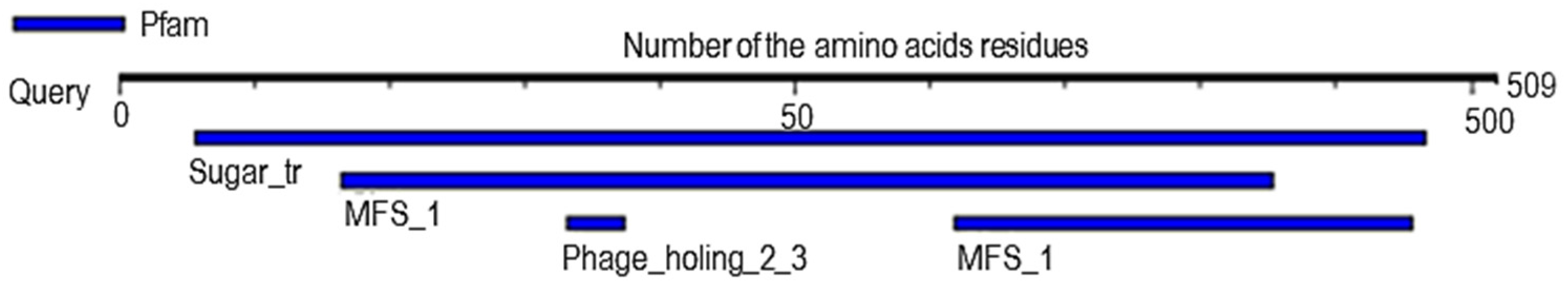
2.6. Analysis of Probable Functional Motifs Presents in Slc2a4 Protein
2.7. The Evaluation and Recognition of Active Sites in Slc2a4 Protein
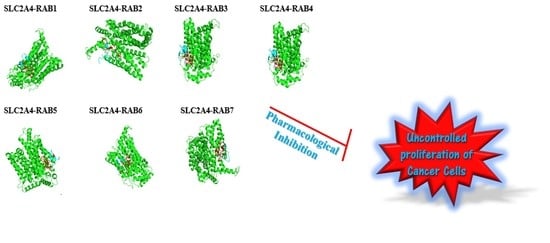
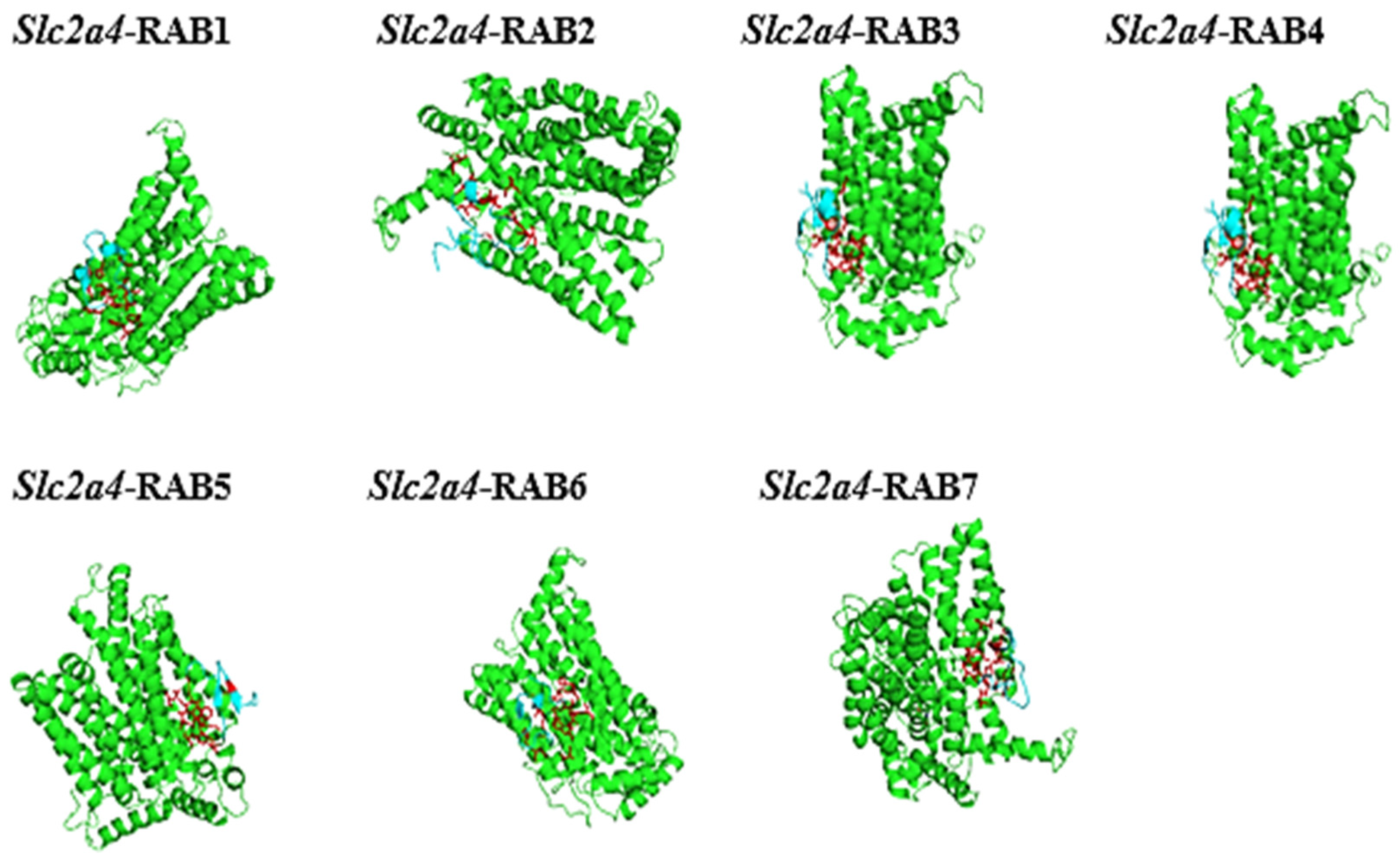
2.8. The Protein-Peptide Interaction between the Putative Anticancer AMPs and Slc2a4 Protein
3. Disccusion
4. Materials and Methods
4.1. Sequence Retrieval and Physicochemical Parameters Analysis of Slc2a4
4.2. Secondary Structure Prediction
4.3. Experimentally Validated Antimicrobial Peptides (AMPs) Data Assessment
4.4. Construction of AMPs Profile Using Hidden Markov Models



Discovery of Novel Putative Anticancer Amps from Genome Sequences

4.5. Determination of the Physicochemical Parameters of the Putative Anti-Anticancer AMPs
4.6. Homology Modelling of the Three-Dimensional Structure of Slc2a4 and Putative Anti-Cancer AMPs
4.7. Evaluation of the Generated Model
4.8. Investigation of the Probable Functional Motif
4.9. Active Site Analysis and Residues Recognition
4.10. In Silico Protein-Protein Interaction Study
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cao, B.; Bray, F.; Beltrán-Sánchez, H.; Ginsburg, O.; Soneji, S.; Soerjomataram, I. Benchmarking life expectancy and cancer mortality: Global comparison with cardiovascular disease 1981–2010. BMJ 2017, 357, j2765. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.N.; Biswas, S.; Reddy, N.D.; Jayashree, B.S.; Kumar, N.; Rao, C.M. In vitro and in vivo anticancer studies of 2′-hydroxy chalcone derivatives exhibit apoptosis in colon cancer cells by HDAC inhibition and cell cycle arrest. EXCLI J. 2017, 16, 448–463. [Google Scholar] [PubMed]
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a Preventable Disease that Requires Major Lifestyle Changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.; Adekola, K.U.; Rosen, S.T.; Shanmugam, M. Cancer metabolism as a therapeutic target. Oncology 2013, 27, 460–467. [Google Scholar] [PubMed]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.W.; Bray, F.; Forman, D.; Ohgaki, H.; Straif, K.; Ullrich, A.; Wild, C.P. Cancer prevention as part of precision medicine: ‘Plenty to be done’. Carcinogenesis 2016, 37, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.J.; Chen, C.; Zhao, Y.; Wang, P.C. Circumventing tumorresistance to chemotherapy by nanotechnology Methods. Mol. Biol. 2010, 596, 467–488. [Google Scholar]
- Desai, A.G.; Qazi, G.N.; Ganju, R.K.; El-Tamer, M.; Singh, J.; Saxena, A.K.; Bedi, Y.S.; Taneja, S.C.; Bhat, H.K. Medicinal plants and cancer chemoprevention. Curr. Drug Metab. 2008, 9, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Wei, C.; Hresko, R.C.; Bajpai, R.; Heitmeier, M.; Matulis, S.M.; Nooka, A.K.; Rosen, S.T.; Hruz, P.W.; Schiltz, G.E.; et al. In silico modeling-based identification of glucose transporter 4 (GLUT4)-selective inhibitors for cancer therapy. J. Biol. Chem. 2015, 290, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Tarazona-Santos, E.; Fabbri, C.; Yeager, M.; Magalhães, W.C.S.; Burdett, L.; Crenshaw, A.; Pettener, D.; Chanock, S.J. Diversity in the glucose transporter-4 gene (SLC2A4) in Humans reflects the action of natural selection along the old-world primates evolution. PLoS ONE 2010, 5, e9827. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.B.; Figueroa, A.; Pulido, E.G.; Campelo, R.G.; Aparicio, L.A. Potential role of sugar transporters in cancer and their relationship with anticancer therapy. Int. J. Endocrinol. 2010, 2010, 205357. [Google Scholar] [CrossRef] [PubMed]
- Oyinloye, B.; Adenowo, F.; Gxaba, N.; Kappo, A. The promise of antimicrobial peptides for treatment of human schistosomiasis. Curr. Drug Targets 2014, 15, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Michelle Furtado, L.; Poon, V.; Klip, A. GLUT4 activation: Thoughts on possible mechanisms. Acta Physiol. Scand. 2003, 178, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Stuart, C.A.; Yin, D.; Howell, M.E.; Dykes, R.J.; Laffan, J.J.; Ferrando, A.A. Hexose transporter mRNAs for GLUT4, GLUT5, and GLUT12 predominate in human muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1067–E1073. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J. Essential Bioinformatics; Cambridge University Press: New York, NY, USA, 2000; p. 352. [Google Scholar]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Pirri, G.; Nicoletto, S. Antimicrobial peptides: An overview of a promising class of therapeutics. Open Life Sci. 2007, 2, 1–33. [Google Scholar] [CrossRef]
- Remaut, H.; Waksman, G. Protein–protein interaction through β-strand addition. Trends Biochem. Sci. 2006, 31, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. In Current Protocols in Bioinformatics; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 5–8. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, G.; Sharma, P.; Anant, A.; Deshmukh, S.; Kaushik, H.; Gopal, K.; Srivastava, N.; Sharma, N.; Garg, L.C. Identification and modeling of a drug target for Clostridium perfringens SM101. Bioinformation 2010, 4, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Roche, D.B.; Brackenridge, D.A.; McGuffin, L.J. Proteins and their interacting partners: An introduction to protein–ligand binding site prediction methods. Int. J. Mol. Sci. 2015, 16, 29829–29842. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, L.M.A.; Villaamil, V.M.; Calvo, M.B.; Rubira, L.V.; Rois, J.M.; Valladares-Ayerbes, M.; Campelo, R.G.; Bolós, M.; Pulido, E.G. Glucose transporter expression and the potential role of fructose in renal cell carcinoma: A correlation with pathological parameters. Mol. Med. Rep. 2010, 3, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Noguchi, Y.; Satoh, S.; Hayashi, H.; Inayama, Y.; Kitamura, H. Expression of facilitative glucose transporter isoforms in lung carcinomas: Its relation to histologic type, differentiation grade, and tumor stage. Mod. Pathol. 1998, 11, 437–443. [Google Scholar] [PubMed]
- Garrido, P.; Osorio, F.G.; Morán, J.; Cabello, E.; Alonso, A.; Freije, J.M.; González, C. Loss of GLUT4 induces metabolic reprogramming and impairs viability of breast cancer cells. J. Cell. Physiol. 2015, 230, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shen, H.; Zhang, L.G.; Liu, J.; Cao, X.G.; Yao, A.L.; Kang, S.S.; Gao, W.X.; Han, H.; Cao, F.H.; et al. Construction and analysis of protein-protein interaction networks based on proteomics data of prostate cancer. Int. J. Mol. Med. 2016, 37, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Hsiao, M. Solute Carrier Family 2 Member 4 Regulates TRIM24-DDX58 Axis to Promote Head and Neck Cancer Metastasis. FASEB J. 2017, 31 (Suppl. 808.5), 808. [Google Scholar]
- De Jesus, A.J.; Allen, T.W. The determinants of hydrophobic mismatch response for transmembrane helices. Biochim. Biophys. Acta 2013, 1828, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Schell, D.; Grimsley, G.R. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Grimsley, G.R.; Scholtz, J.M. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J. Biol. Chem. 2009, 284, 13285–13289. [Google Scholar] [CrossRef] [PubMed]
- Petsko, G.A.; Ringe, D. Protein Structure and Function; New Science Press: London, UK, 2004. [Google Scholar]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Nowick, J.S. Exploring β-sheet structure and interactions with chemical model systems. Acc. Chem. Res. 2008, 41, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Gujjula, K.R. Prediction and Comparison of HIV-1 Protease Inhibitor Binding Energies by Various Molecular Docking Methods. Ph.D. Dissertation, National Institute of Technology Rourkela, Orissa, India, 2008. [Google Scholar]
- The UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Ikai, A.J. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://pymol.Org (accessed on 25 September 2016).
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. Lect. Notes Comput. Sci. 2002, 2452, 185–200. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ProtParam Parameters | Values |
|---|---|
| Number of amino acids | 509 |
| Molecular weight | 54,895.50 Da |
| Theoretical Pi | 6.86 |
| Atomic composition | Carbon C: 2530 Hydrogen H: 4009 Nitrogen N: 641 Oxygen O: 686 Sulfur S: 16 |
| Formula | C2530H4009N641O686S16 |
| Number of negatively charged residues | 33 |
| Number of positively charged residues | 33 |
| Extinction coefficient | 59,610 Abs 0.1% (=1 g/L) 1.086, assuming all pairs of Cys residues form cystines 59,360 Abs 0.1% (=1 g/L) 1.081, assuming all Cys residues are reduced |
| Total number of atoms | 7882 |
| Estimated Half-life | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo). |
| Aliphatic index | 118.61 |
| Grand average of hydropathicity (GRAVY) | 0.556 |
| Instability index | 39.83 |
| Putative AMPs | Mass (Da) | Most Common Amino Acids | Isoelectric Point | Net Charge | Total Hydrophobic Ratio (%) | Bosman Index (kcal/mol) | Half-Life (h) | Sequence Similarity with Other Molecules and Percentage (%) |
|---|---|---|---|---|---|---|---|---|
| RAB 1 | 3038.03 | Cys: 20.69 | 6.03 | 0 | 51 | 0.18 | 30 | AP02325: 75 |
| RAB 2 | 3091.14 | Ile: 16.67 | 6.03 | 0 | 50 | −0.11 | 30 | AP01777: 80.64 |
| RAB 3 | 3313.15 | Ile/Val: 12.9 | 7.76 | 2 | 51 | 0.48 | 30 | AP01990: 74.19 |
| RAB 4 | 3245.94 | Val: 12.9 | 5.53 | 0 | 51 | 0.56 | 30 | AP01062: 77.41 |
| RAB 5 | 3156.35 | Cys: 21.43 | 4.31 | −1 | 46 | 1.00 | 30 | AP00274: 59.37 |
| RAB 6 | 2930.31 | Cys: 21.43 | 3.85 | −1 | 60 | −1.00 | 30 | AP01777: 56.66 |
| RAB 7 | 3197.90 | Cys: 20.69 | 8.38 | 3 | 51 | 3 | 30 | AP02661: 61.29 |
| Putative Anti-Cancer AMPs | C-Score | Exp. TM Score | Exp. RMSD (Å) |
|---|---|---|---|
| RAB 1 | 0.81 | 0.82 ± 0.08 | 0.5 ± 0.5 |
| RAB 2 | 0.71 | 0.87 ± 0.09 | 0.5 ± 0.5 |
| RAB 3 | 0.70 | 0.81 ± 0.09 | 0.6 ± 0.6 |
| RAB 4 | 0.71 | 0.81 ± 0.09 | 0.6 ± 0.6 |
| RAB 5 | 0.69 | 0.81 ± 0.09 | 0.5 ± 0.5 |
| RAB 6 | 0.44 | 0.77 ± 0.10 | 0.9 ± 0.9 |
| RAB 7 | 0.67 | 0.80 ± 0.09 | 0.6 ± 0.6 |
| Slc2a4 | 0.42 | 0.77 ± 0.10 | 6.4 ± 3.9 |
| S/N | Pfam ID | Position | E-Value | Description |
|---|---|---|---|---|
| 1 | Sugar_tr | 28–483 | 1.8 × 10−152 | Sugar (and other) transporter |
| 2 | MFS_1 | 82–427 309–478 | 1.8 × 10−15 4 × 10−3 | Major Facilitator Superfamily |
| 3 | Phage_holin_2_3 | 166–187 | 7.5 × 10−2 | Bacteriophage holin family HP1 |
| Name of Server | Name of Ligand | Residue Number | C-Score |
|---|---|---|---|
| COACH SERVER | Maltose (MAL) | 42, 46, 177, 180, 181, 184, 298, 299, 304, 333, 395, 396, 404, 431 | 0.19 |
| Cytochalasin B (5RH) | 96, 153, 177, 180, 298, 304, 400, 424, 427, 428, 431 | 0.10 | |
| Cholesterol hemisuccinate (Y01) | 97, 275, 292, 296, 300, 426, 429, 434 | 0.09 | |
| (2~{S})-3-(2-bromophenyl)-2-[2-(4-methoxyphenyl)ethanoylamino]-~{N}-[(1~{S})-1-phenylethyl] propenamide (5RF) | 38, 96, 153, 176, 298, 395, 396, 420, 424, 427, 428 | 0.05 | |
| Octyl Glucose Neopentyl Glycol (37X) | 22, 23, 26, 218, 219, 222, 246 | 0.03 | |
| (2S)-2,3Dihydroxypropyl (7Z)-pentadec-7-enoate (78M) | 90, 91, 93, 94, 142, 426, 427 | 0.02 | |
| Octyl Glucose Neopentyl Glycol (37X) | 94, 97, 98, 101, 118, 143 | 0.02 | |
| methyl-α-d-glucopyranoside (GYP) | 296, 430, 433, 434, 452, 453, 456, 457 | 0.02 | |
| N-[(1R)-1-phosphonoethyl]-l-alaninamide (AFS) | 39, 43, 177, 180, 181, 184, 329, 333 | 0.02 | |
| M-cresol (CRS) | 306, 309 | - | |
| TM-Site Results | Maltose (MAL) (2), β-d-glucose (BGC) (2), 6-bromo-6-deoxy-β-d-glucopyranose (6BG) (1) | 42, 46, 177, 180, 181, 184, 298, 299, 304, 333, 395, 396, 404, 431 | 0.4 |
| 5RH (1), (2~{S})-3-(2-bromophenyl)-2-[2-(4-methoxyphenyl)ethanoylamino]-~{N}-[(1~{S})-1-phenylethyl] propenamide (5RF)(1), (2~{S})-3-(4-fluorophenyl)-2-[2-(3-hydroxyphenyl)ethanoylamino]-~{N}-[(1~{S})-1-phenylethyl]propenamide(5RE(1) | 96, 153, 177, 180, 298, 304, 400, 424, 427, 428, 431 | 0.2 | |
| Cholesterol hemisuccinate (Y01) (2) | 97, 275, 292, 296, 300, 426, 429, 434 | 0.1 | |
| Octyl Glucose Neopentyl Glycol (37X) (1) | 22, 23, 26, 218, 219, 222, 246 | 0.1 | |
| S-Site Results | 2′-deoxycytidine-5′-monophosphate (DCM) (1), 2′-deoxyuridine 5′-monophosphate (UMP) (1) | 349, 350 | 0.16 |
| M-cresol (CRS) (1), adenosine-5′-phosphosulfate (ADX) (1) | 304, 306, 309 | 0.15 | |
| magnesium ion (Mg2+) (1), calcium ion (Ca2+) (1) | 224, 225, 226, 228 | 0.15 | |
| magnesium ion (Mg2+) (1) | 162, 165 | 0.14 | |
| l-α-glycerophosphorylethanol amine (GPE) (1), Fe (II) ion (Fe2+) (1) | 239, 299, 302, 303, 400, 409 | 0.14 |
| Molecule (AMPs) | Binding Affinity for Geometry Scores | Global Energy (Binding Energy for the Solution) (kcal/mol) |
|---|---|---|
| RAB 1 | 12,392 | −39.13 |
| RAB 2 | 11,528 | −36.74 |
| RAB 3 | 10,154 | −53.99 |
| RAB 4 | 10,768 | −55.01 |
| RAB 5 | 11,146 | −68.19 |
| RAB 6 | 10,556 | −25.85 |
| RAB 7 | 11,558 | −36.88 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aruleba, R.T.; Adekiya, T.A.; Oyinloye, B.E.; Kappo, A.P. Structural Studies of Predicted Ligand Binding Sites and Molecular Docking Analysis of Slc2a4 as a Therapeutic Target for the Treatment of Cancer. Int. J. Mol. Sci. 2018, 19, 386. https://doi.org/10.3390/ijms19020386
Aruleba RT, Adekiya TA, Oyinloye BE, Kappo AP. Structural Studies of Predicted Ligand Binding Sites and Molecular Docking Analysis of Slc2a4 as a Therapeutic Target for the Treatment of Cancer. International Journal of Molecular Sciences. 2018; 19(2):386. https://doi.org/10.3390/ijms19020386
Chicago/Turabian StyleAruleba, Raphael Taiwo, Tayo Alex Adekiya, Babatunji Emmanuel Oyinloye, and Abidemi Paul Kappo. 2018. "Structural Studies of Predicted Ligand Binding Sites and Molecular Docking Analysis of Slc2a4 as a Therapeutic Target for the Treatment of Cancer" International Journal of Molecular Sciences 19, no. 2: 386. https://doi.org/10.3390/ijms19020386
APA StyleAruleba, R. T., Adekiya, T. A., Oyinloye, B. E., & Kappo, A. P. (2018). Structural Studies of Predicted Ligand Binding Sites and Molecular Docking Analysis of Slc2a4 as a Therapeutic Target for the Treatment of Cancer. International Journal of Molecular Sciences, 19(2), 386. https://doi.org/10.3390/ijms19020386