Molecular Understanding of the Activation of CB1 and Blockade of TRPV1 Receptors: Implications for Novel Treatment Strategies in Osteoarthritis



Abstract
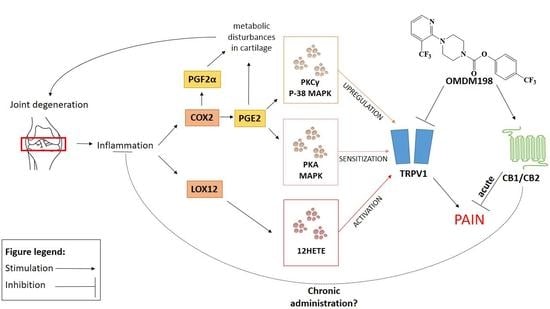
1. Introduction
2. Results
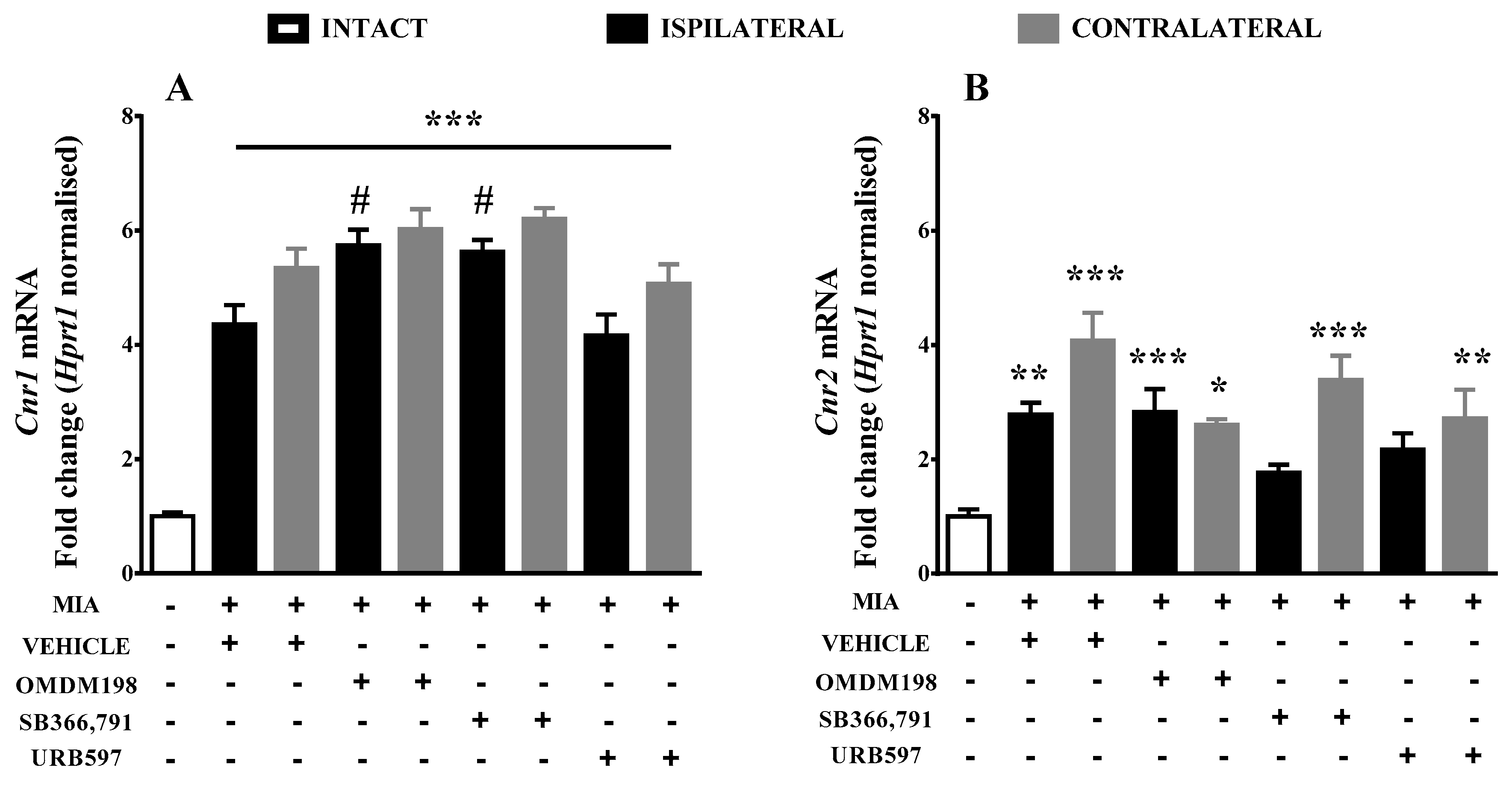
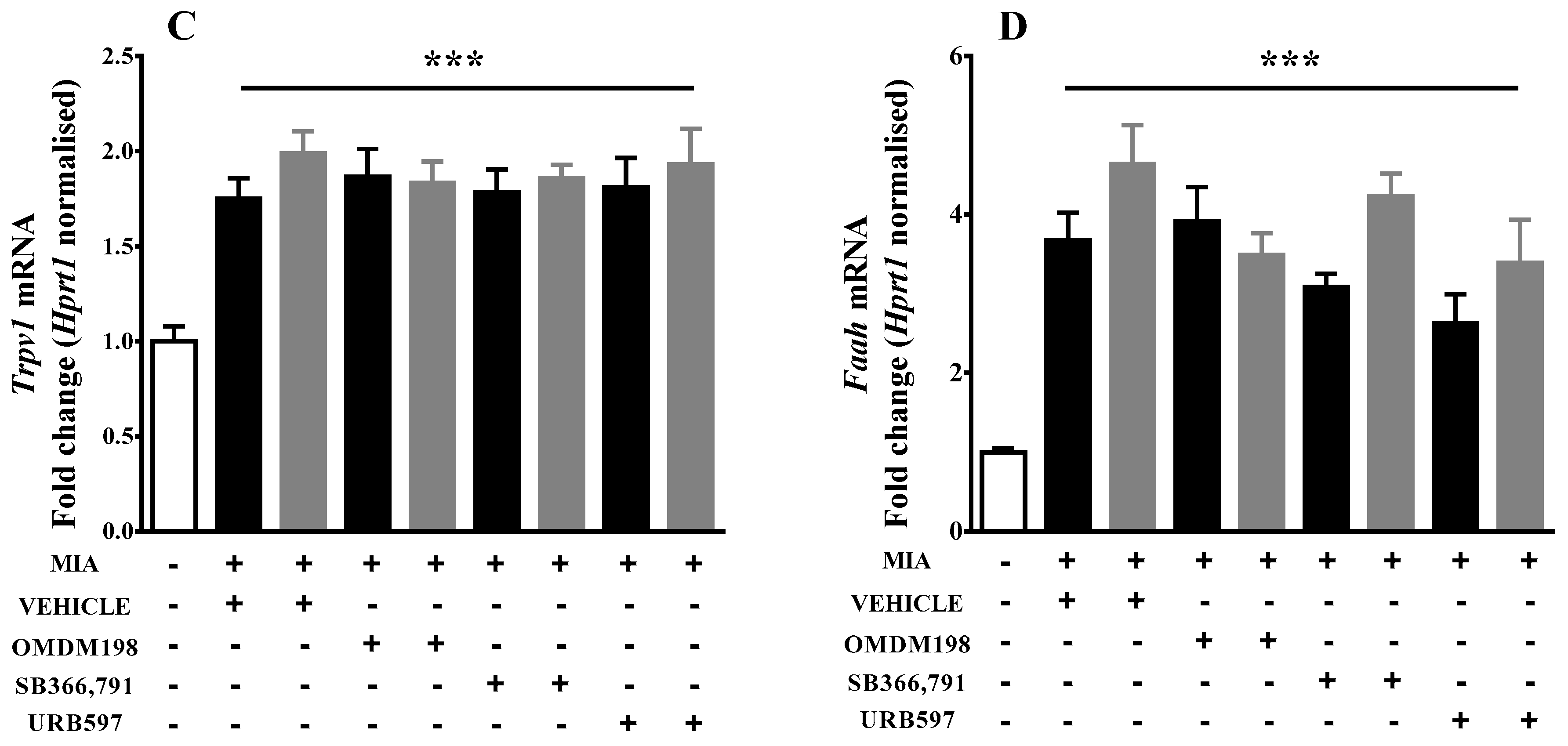
2.1. Changes in Cnr1, Cnr2, Faah, and Trpv1 Gene Expression in the Lumbar Spinal Cord
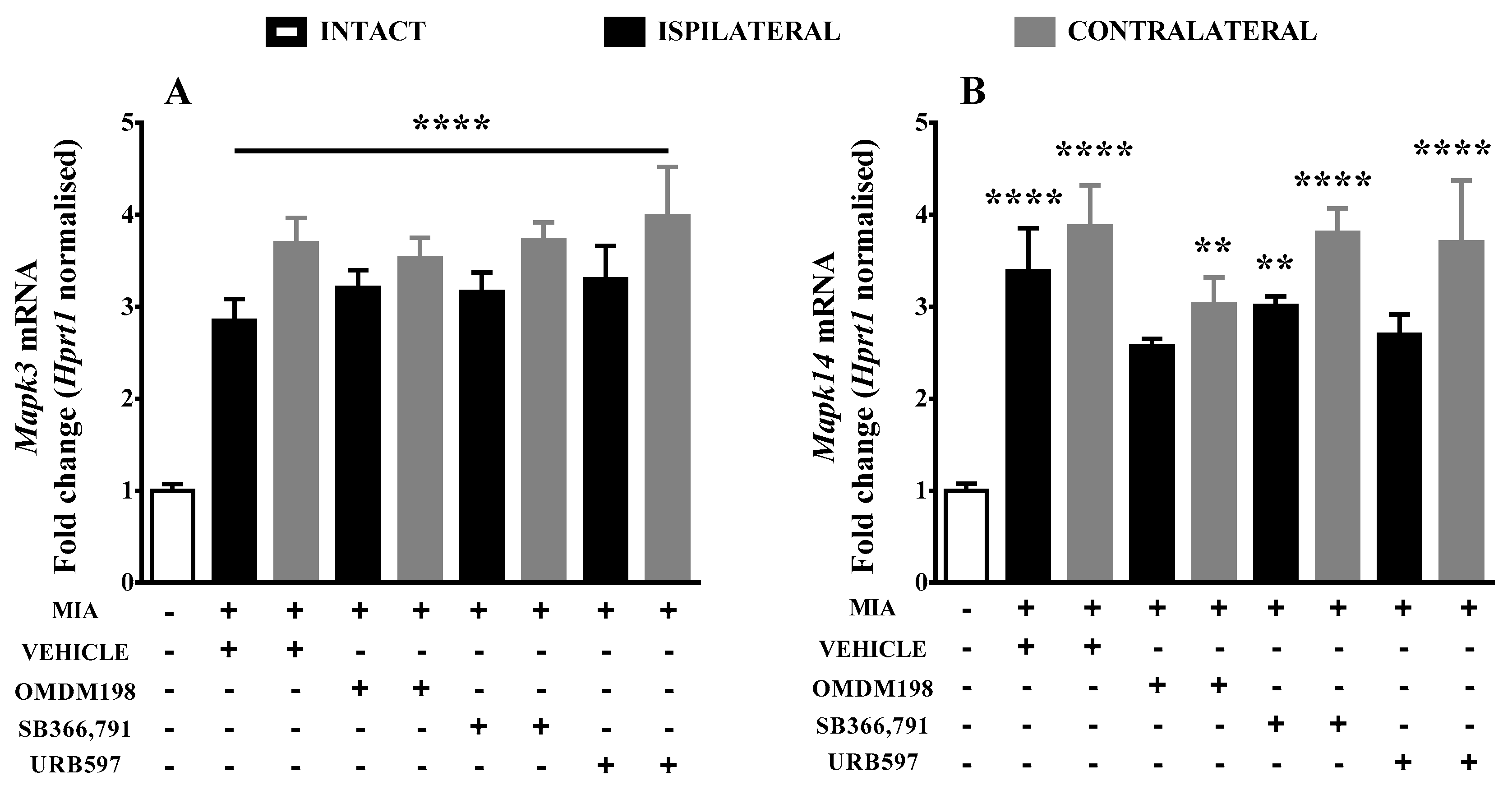
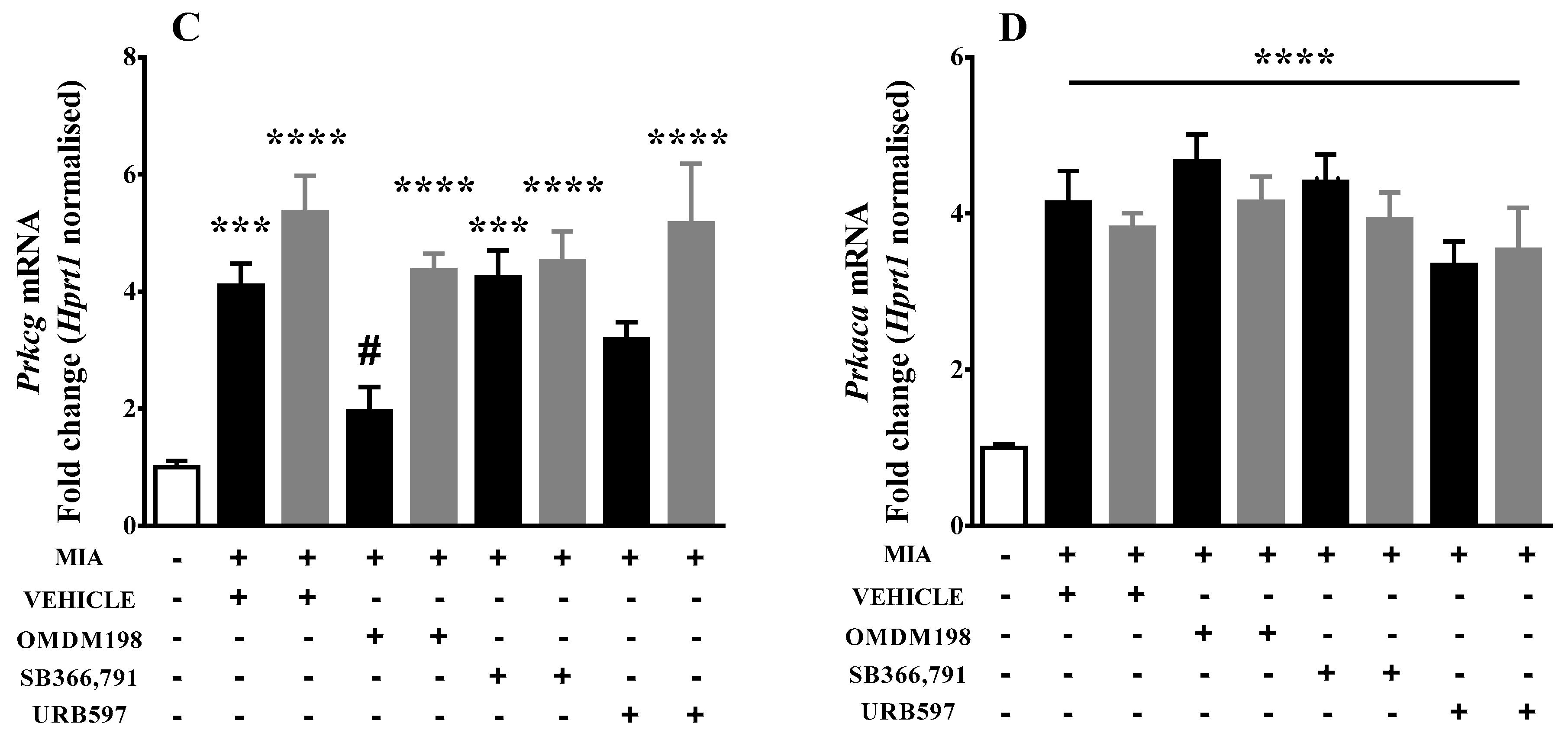
2.2. Changes in Gene Expression of Kinases Involved in TRPV1 Sensitization in the Lumbar Spinal Cord
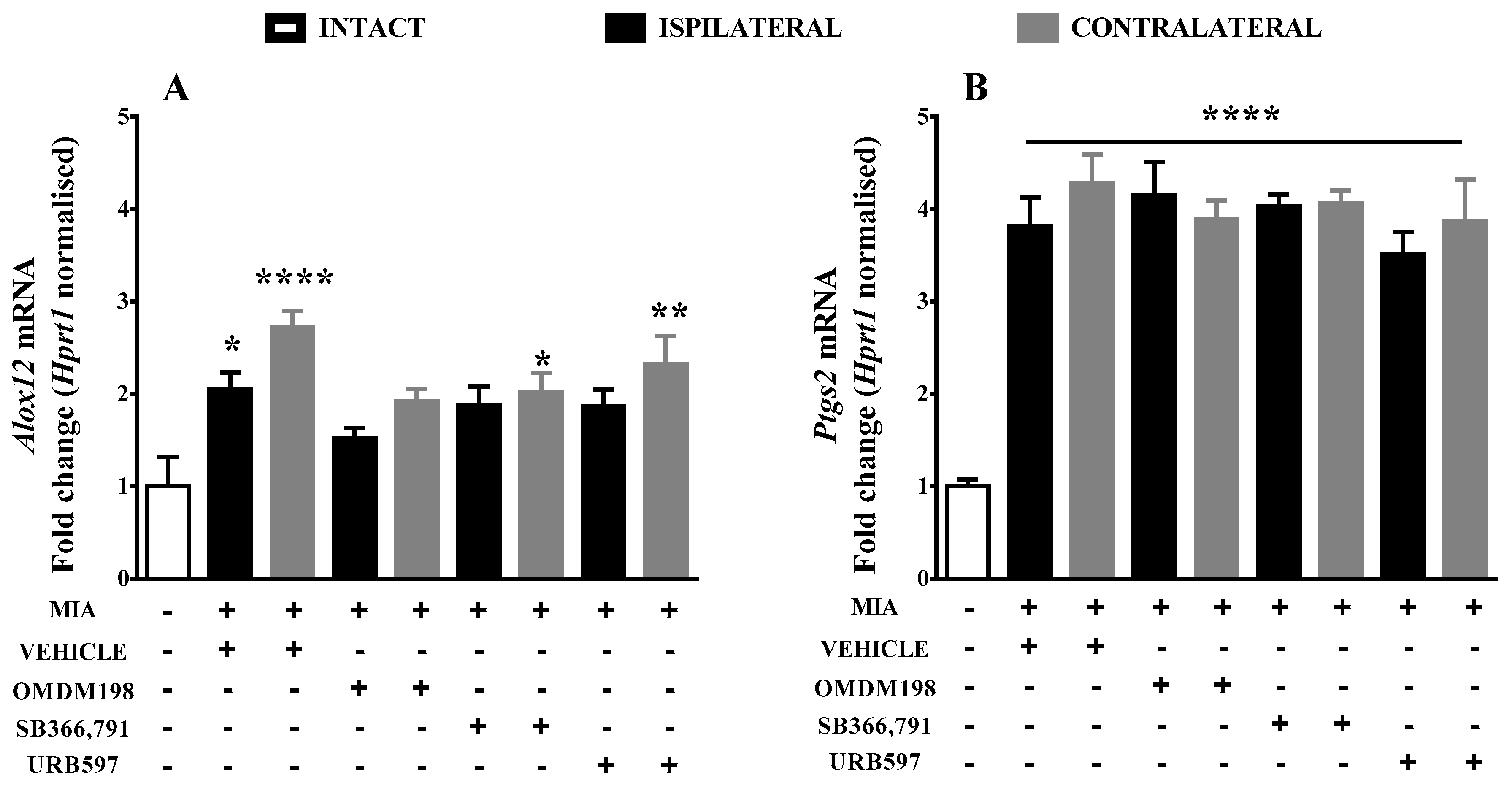
2.3. Changes of the AEA Alternative Degrading Enzymes Gene Expression in the Lumbar Spinal Cord
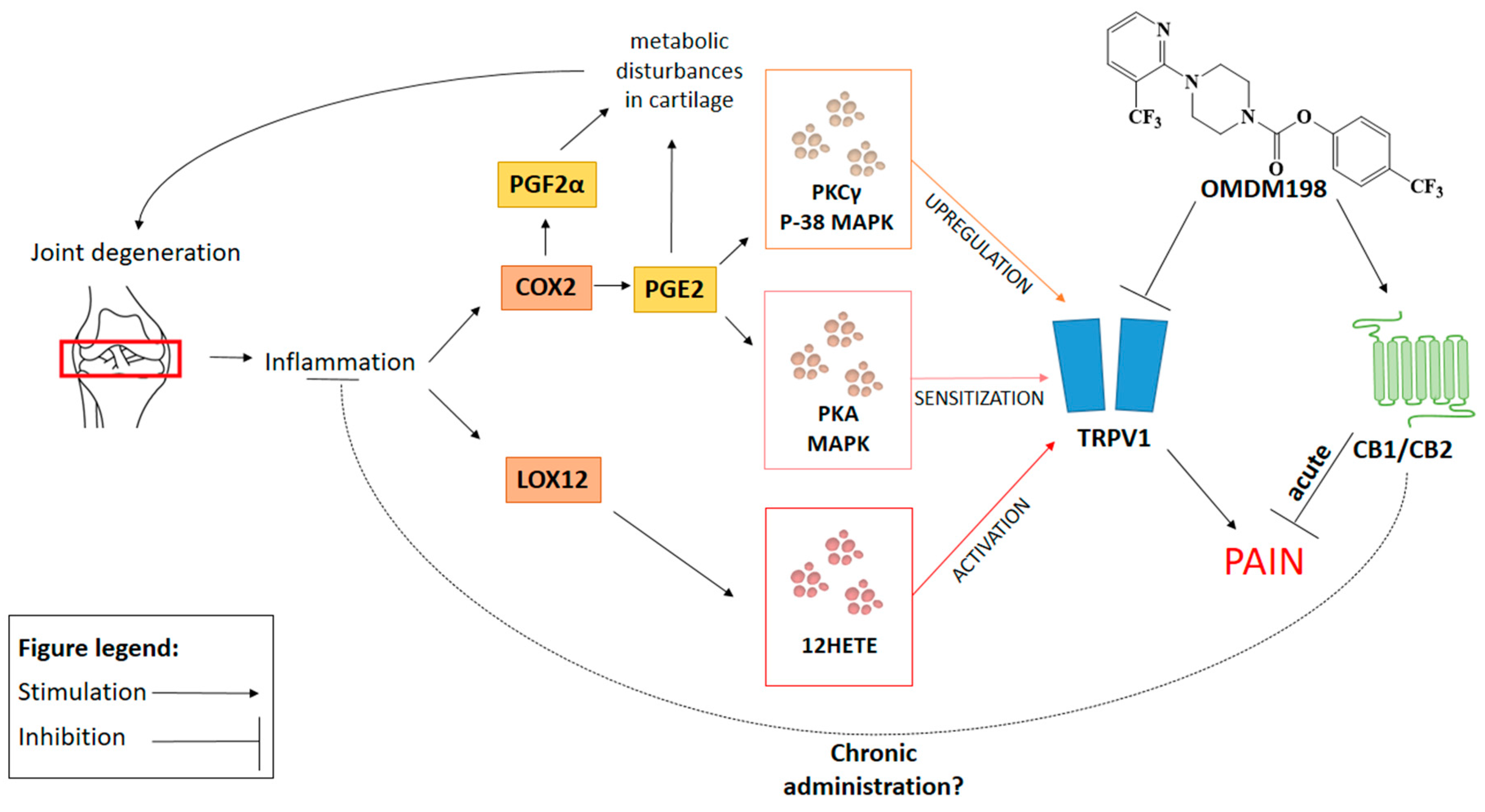
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs and Reagents
4.3. OA Induction
4.4. Treatment Paradigm
4.5. Tissue Isolation
4.6. RNA Preparation
4.7. Quantitative Polymerase Chain Reaction (qPCR)
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonylglycerol |
| AEA | Anandamide |
| cAMP | Cyclic adenosine monophosphate |
| CB1 | Cannabinoid receptor type 1 |
| CB2 | Cannabinoid receptor type 2 |
| COX2 | Cyclooxygenase-2 |
| CT | Threshold cycle |
| DMSO | Dimethyl sulfoxide |
| DRGs | Dorsal root ganglions |
| ECS | Endocannabinoid system |
| FAAH | Fatty acid amide hydrolase |
| HETE | Hydroxyeicosatetraenoic acid |
| HPETE | Hydroperoxyeicosatetraenoic acid |
| MAPK | Mitogen-activated protein kinases |
| MIA | Monoiodoacetate |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| OA | Osteoarthritis |
| qPCR | Quantitive polymerase chain reaction |
| PGE2 | Prostaglandine E2 |
| PGF2α | Prostamide F2α |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| TRPV1 | Transient receptor potential cation channel subfamily V member 1 |
References
- Haseeb, A.; Haqqi, T.M. Immunopathogenesis of osteoarthritis. Clin. Immunol. 2013, 146, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kidd, B. Mechanisms of pain in osteoarthritis. HSS J. 2012, 8, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Lluch, E.; Torres, R.; Nijs, J.; van Oosterwijck, J. Evidence for central sensitization in patients with osteoarthritis pain: A systematic literature review. Eur. J. Pain 2014, 18, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Van Walsem, A.; Pandhi, S.; Nixon, R.M.; Guyot, P.; Karabis, A.; Moore, R.A. Relative benefit-risk comparing diclofenac to other traditional non-steroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors in patients with osteoarthritis or rheumatoid arthritis: A network meta-analysis. Arthritis Res. Ther. 2015, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.; Ahmad, K.; Rice, A.S.C. The synthetic cannabinoid WIN55,212-2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Br. J. Pharmacol. 2001, 133, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Schuelert, N.; McDougall, J.J. Cannabinoid-mediated antinociception is enhanced in rat osteoarthritic knees. Arthritis Rheum. 2008, 58, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Schuelert, N.; Johnson, M.P.; Oskins, J.L.; Jassal, K.; Chambers, M.G.; McDougall, J.J. Local application of the endocannabinoid hydrolysis inhibitor URB597 reduces nociception in spontaneous and chemically induced models of osteoarthritis. Pain 2011, 152, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.K.; Neufeld, M.; Jesus, C.H.A.; Cunha, J.M. Peripheral antinociceptive effect of anandamide and drugs that affect the endocannabinoid system on the formalin test in normal and streptozotocin-diabetic rats. Neuropharmacology 2012, 63, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Danovitch, I.; Gorelick, D.A. State of the art treatments for cannabis dependence. Psychiatr. Clin. N. Am. 2012, 35, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kinsey, S.G.; Liu, Q.S.; Hruba, L.; McMahon, L.R.; Grim, T.W.; Merritt, C.R.; Wise, L.E.; Abdullah, R.A.; Selley, D.E.; et al. Full Fatty Acid Amide Hydrolase Inhibition Combined with Partial Monoacylglycerol Lipase Inhibition: Augmented and Sustained Antinociceptive Effects with Reduced Cannabimimetic Side Effects in Mice. J. Pharmacol. Exp. Ther. 2015, 354, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Sagar, D.R.; Staniaszek, L.E.; Okine, B.N.; Woodhams, S.; Norris, L.M.; Pearson, R.G.; Garle, M.J.; Alexander, S.P.H.; Bennett, A.J.; Barrett, D.A.; et al. Tonic modulation of spinal hyperexcitability by the endocannabinoid receptor system in a rat model of osteoarthritis pain. Arthritis Rheum. 2010, 62, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, F.; di Marzo, V. “Redundancy” of endocannabinoid inactivation: New challenges and opportunities for pain control. ACS Chem. Neurosci. 2012, 3, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; de Petrocellis, L. Why do cannabinoid receptors have more than one endogenous ligand? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3216–3228. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, J.; Urban, L.; Bevan, S.; Nagy, I. Anandamide regulates neuropeptide release from capsaicin-sensitive primary sensory neurons by activating both the cannabinoid 1 receptor and the vanilloid receptor 1 in vitro. Eur. J. Neurosci. 2003, 17, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; de Petrocellis, L.; Pryce, G.; Baker, D.; Guglielmotti, V.; di Marzo, V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 2006, 139, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Starowicz, K.; Makuch, W.; Korostynski, M.; Malek, N.; Slezak, M.; Zychowska, M.; Petrosino, S.; de Petrocellis, L.; Cristino, L.; Przewlocka, B.; et al. Full Inhibition of Spinal FAAH Leads to TRPV1-Mediated Analgesic Effects in Neuropathic Rats and Possible Lipoxygenase-Mediated Remodeling of Anandamide Metabolism. PLoS ONE 2013, 8, e60040. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Otto, W.R.; Sanchez-Herrera, D.; Facer, P.; Yiangou, Y.; Korchev, Y.; Birch, R.; Benham, C.; Bountra, C.; Chessell, I.P.; et al. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain 2008, 138, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Hermann, H.; de Petrocellis, L.; Bisogno, T.; Schiano Moriello, A.; Lutz, B.; di Marzo, V. Dual effect of cannabinoid CB1 receptor stimulation on a vanilloid VR1 receptor-mediated response. Cell. Mol. Life Sci. 2003, 60, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Bhave, G.; Zhu, W.; Wang, H.; Brasier, D.J.; Oxford, G.S.; Gereau, R.W. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 2002, 35, 721–731. [Google Scholar] [CrossRef]
- Rathee, P.K.; Distler, C.; Obreja, O.; Neuhuber, W.; Wang, G.K.; Wang, S.-Y.; Nau, C.; Kress, M. PKA/AKAP/VR-1 module: A common link of Gs-mediated signaling to thermal hyperalgesia. J. Neurosci. 2002, 22, 4740–4745. [Google Scholar]
- Maione, S.; Bisogno, T.; de Novellis, V.; Palazzo, E.; Cristino, L.; Valenti, M.; Petrosino, S.; Guglielmotti, V.; Rossi, F.; di Marzo, V. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J. Pharmacol. Exp. Ther. 2006, 316, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Starowicz, K.; Maione, S.; Cristino, L.; Palazzo, E.; Marabese, I.; Rossi, F.; de Novellis, V.; di Marzo, V. Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J. Neurosci. 2007, 27, 13739–13749. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Pajak, A.; Kolosowska, N.; Kucharczyk, M.; Starowicz, K. The importance of TRPV1-sensitisation factors for the development of neuropathic pain. Mol. Cell. Neurosci. 2015, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef] [PubMed]
- Olah, Z.; Karai, L.; Iadarola, M.J. Anandamide activates vanilloid receptor 1 (VR1) at acidic pH in dorsal root ganglia neurons and cells ectopically expressing VR1. J. Biol. Chem. 2001, 276, 31163–31170. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Mrugala, M.; Makuch, W.; Kolosowska, N.; Przewlocka, B.; Binkowski, M.; Czaja, M.; Morera, E.; di Marzo, V.; Starowicz, K. A multi-target approach for pain treatment: Dual inhibition of fatty acid amide hydrolase and TRPV1 in a rat model of osteoarthritis. Pain 2015, 156, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, K.A.; Kerr, B.J. The transition from acute to chronic pain: Understanding how different biological systems interact. Can. J. Anesth. 2014, 61, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.; Sung, B.; Ji, R.-R.; Mao, J. Upregulation of spinal cannabinoid-1-receptors following nerve injury enhances the effects of Win 55,212-2 on neuropathic pain behaviors in rats. Pain 2003, 105, 275–283. [Google Scholar] [CrossRef]
- Burston, J.J.; Sagar, D.R.; Shao, P.; Bai, M.; King, E.; Brailsford, L.; Turner, J.M.; Hathway, G.J.; Bennett, A.J.; Walsh, D.A.; et al. Cannabinoid CB2 Receptors Regulate Central Sensitization and Pain Responses Associated with Osteoarthritis of the Knee Joint. PLoS ONE 2013, 8, e80440. [Google Scholar] [CrossRef] [PubMed]
- Grim, T.W.; Morales, A.J.; Gonek, M.M.; Wiley, J.L.; Thomas, B.F.; Endres, G.W.; Sim-Selley, L.J.; Selley, D.E.; Negus, S.S.; Lichtman, A.H. Stratification of Cannabinoid 1 Receptor (CB1R) Agonist Efficacy: Manipulation of CB1R Density through Use of Transgenic Mice Reveals Congruence between In Vivo and In Vitro Assays. J. Pharmacol. Exp. Ther. 2016, 359, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Nakazato, E.; Fujiuchi, A.; Hara, T.; Imai, A. Involvement of an increased spinal TRPV1 sensitization through its up-regulation in mechanical allodynia of CCI rats. Neuropharmacology 2005, 49, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Samad, T.A.; Jin, S.-X.; Schmoll, R.; Woolf, C.J. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 2002, 36, 57–68. [Google Scholar] [CrossRef]
- Obata, K.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Mizushima, T.; Katsura, H.; Fukuoka, T.; Tokunaga, A.; Noguchi, K. Role of Mitogen-Activated Protein Kinase Activation in Injured and Intact Primary Afferent Neurons for Mechanical and Heat Hypersensitivity after Spinal Nerve Ligation. J. Neurosci. 2004, 24, 10211–10222. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; St-Jacques, B.; Rudakou, U.; Kim, Y.N. Stimulating TRPV1 externalization and synthesis in dorsal root ganglion neurons contributes to PGE2 potentiation of TRPV1 activity and nociceptor sensitization. Eur. J. Pain 2017, 21, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Koda, K.; Hyakkoku, K.; Ogawa, K.; Takasu, K.; Imai, S.; Sakurai, Y.; Fujita, M.; Ono, H.; Yamamoto, M.; Fukuda, I.; et al. Sensitization of TRPV1 by protein kinase C in rats with mono-iodoacetate-induced joint pain. Osteoarthr. Cartil. 2016, 24, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Fitzsimmons, B.; Mohan, A.; Zhang, L.; Terrando, N.; Kordasiewicz, H.; Ji, R.-R. Intrathecal administration of antisense oligonucleotide against p38α but not p38β MAP kinase isoform reduces neuropathic and postoperative pain and TLR4-induced pain in male mice. Brain. Behav. Immun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Miletic, G.; Hermes, J.L.; Bosscher, G.L.; Meier, B.M.; Miletic, V. Protein kinase C γ-mediated phosphorylation of GluA1 in the postsynaptic density of spinal dorsal horn neurons accompanies neuropathic pain, and dephosphorylation by calcineurin is associated with prolonged analgesia. Pain 2015, 156, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Chapman, R.J.; Woodhams, S.; Sagar, D.R.; Turner, J.; Burston, J.J.; Bullock, C.; Paton, K.; Huang, J.; Wong, A.; et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann. Rheum. Dis. 2015, 74, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Kozak, K.R.; Prusakiewicz, J.J.; Marnett, L.J. Oxidative metabolism of endocannabinoids by COX-2. Curr. Pharm. Des. 2004, 10, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gatta, L.; Piscitelli, F.; Giordano, C.; Boccella, S.; Lichtman, A.; Maione, S.; Di Marzo, V. Discovery of Prostamide F2α and Its Role in Inflammatory Pain and Dorsal Horn Nociceptive Neuron Hyperexcitability. PLoS ONE 2012, 7, e31111. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Whiteman, M.; Mattey, D.L.; Halliwell, B. Raised levels of F(2)-isoprostanes and prostaglandin F(2α) in different rheumatic diseases. Ann. Rheum. Dis. 2001, 60, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F2α receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-β. Nat. Med. 2009, 15, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Telleria-Diaz, A.; Schmidt, M.; Kreusch, S.; Neubert, A.-K.; Schache, F.; Vazquez, E.; Vanegas, H.; Schaible, H.-G.; Ebersberger, A. Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: Involvement of prostaglandins and endocannabinoids. Pain 2010, 148, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Staniaszek, L.; Norris, L.; Kendall, D.; Barrett, D.; Chapman, V. Effects of COX-2 inhibition on spinal nociception: The role of endocannabinoids. Br. J. Pharmacol. 2010, 160, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Favia, A.D.; Convertino, M.; de Vivo, M. The Molecular Basis for Dual Fatty Acid Amide Hydrolase (FAAH)/Cyclooxygenase (COX) Inhibition. ChemMedChem 2016, 11, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- De Lange-Brokaar, B.J.E.; Ioan-Facsinay, A.; van Osch, G.J.V.M.; Zuurmond, A.-M.; Schoones, J.; Toes, R.E.M.; Huizinga, T.W.J.; Kloppenburg, M. Synovial inflammation, immune cells and their cytokines in osteoarthritis: A review. Osteoarthr. Cartil. 2012, 20, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Morera, E.; De Petrocellis, L.; Morera, L.; Moriello, A.S.; Ligresti, A.; Nalli, M.; Woodward, D.F.; Di Marzo, V.; Ortar, G. Synthesis and biological evaluation of piperazinyl carbamates and ureas as fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channel dual ligands. Bioorg. Med. Chem. Lett. 2009, 19, 6806–6809. [Google Scholar] [CrossRef] [PubMed]
- Guingamp, C.; Gegout-Pottie, P.; Philippe, L.; Terlain, B.; Netter, P.; Gillet, P. Mono-iodoacetate-induced experimental osteoarthritis: A dose-response study of loss of mobility, morphology, and biochemistry. Arthritis Rheum. 1997, 40, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Chomzynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ipsi | Vehicle | OMDM198 | SB366791 | URB597 |
|---|---|---|---|---|
| Cnr1 | 4.36 ± 0.33 (10) ***↑ | 5.73934 ± 0.28 (10) ***,#↑↑ | 5.62706 ± 0.21 (10) ***,#↑↑ | 4.16213 ± 0.37 (7) *** |
| Cnr2 | 2.77949 ± 0.21 (9) **↑ | 2.829189 ± 0.4 (10) ***↑ | 1.76802 ± 0.14 (9) | 2.166964 ± 0.29 (7)↑ |
| Trpv1 | 1.75039 ± 0.11 (9) ***↑ | 1.86664 ± 0.15 (10) ***↑ | 1.78189 ± 0.12 (10) ***↑ | 1.80975 ± 0.15 (6) *** |
| Faah | 3.67581 ± 0.35 (10) ***↑ | 3.91468 ± 0.43 (10) ***↑ | 3.08223 ± 0.17 (9) ***↑ | 2.63209 ± 0.36 (7) ***↑ |
| Mapk3 | 2.847 ± 0.23 (10) ****↑ | 3.20392 ± 0.19 (10) ****↑ | 3.16103 ± 0.21(10) ****↑ | 3.30138 ± 0.36 (7) ****↑ |
| Mapk14 | 3.38744 ± 0.46 (10) ****↑ | 2.56705 ± 0.08 (8) | 3.01161 ± 0.1 (8) **↑ | 2.69419 ± 0.22 (6)↑ |
| Prkaca | 4.13981 ± 0.4 (10) ****↑ | 4.66819 ± 0.35 (6) ****↑ | 4.40495 ± 0.35 (6) ****↑ | 3.33948 ± 0.3 (7) **** |
| Prkcg | 4.10269 ± 0.37 (10) ***↑ | 1.95838 ± 0.41 (9) #↓ | 4.25047 ± 0.45 (6) ***↑ | 3.18495 ± 0.3 (7)↑ |
| alox12 | 2.045551 ± 0.18 (10) *↑ | 1.519128 ± 0.11 (10) | 1.876361 ± 0.2 (10) | 1.866132 ± 0.18 (7) |
| Ptgs2 | 3.81417 ± 0.31 (10) ****↑ | 4.15244 ± 0.36 (10) ****↑ | 4.03079 ± 0.13 (10) ****↑ | 3.5141 ± 0.24 (7) ****↑ |
| Contra | Vehicle | OMDM198 | SB366791 | URB597 |
|---|---|---|---|---|
| Cnr1 | 5.34396 ± 0.34 (8) ***↑ | 6.02753 ± 0.35 (10)***↑ | 6.20558 ± 0.19 (10) ***↑ | 5.0704 ± 0.34 (8) ***↑ |
| Cnr2 | 4.075169 ± 0.49 (8) ***↑ | 2.604934 ± 0.1 (8) *↑ | 3.388387 ± 0.42 (10) ***↑ | 2.717181 ± 0.5 (9) **↑ |
| Trpv1 | 1.98928 ± 0.12 (8) ***↑ | 1.8344 ± 0.11 (10) ***↑ | 1.85948 ± 0.07 (9) ***↑ | 1.9315 ± 0.19 (9) ***↑ |
| Faah | 4.63649 ± 0.49 (8) ***↑ | 3.49026 ± 0.27 (9) ***↑ | 4.23487 ± 0.28 (10) ***↑ | 3.38967 ± 0.55 (9) ***↑ |
| Mapk3 | 3.69186 ± 0.28 (8) ****↑ | 3.52777 ± 0.22 (9) ****↑ | 3.72732 ± 0.19 (10) ****↑ | 3.98384 ± 0.54 (8) ****↑ |
| Mapk14 | 3.87233 ± 0.45 (8) ****↑ | 3.02531 ± 0.29 (10) **↑ | 3.80339 ± 0.27 (10) ****↑ | 3.69974 ± 0.67 (9) ****↑ |
| Prkaca | 3.81766 ± 0.19 (7) ****↑ | 4.14995 ± 0.32 (8) ****↑ | 3.92648 ± 0.34 (10) ****↑ | 3.53792 ± 0.53 (9) ****↑ |
| Prkcg | 5.34829 ± 0.63 (8) ****↑ | 4.36753 ± 0.28 (9) ****↑ | 4.52286 ± 0.5 (10) ****↑ | 5.16141 ± 1.02 (9) ****↑ |
| alox12 | 2.722801 ± 0.17 (8) ****↑ | 1.917924 ± 0.13 (9) | 2.020144 ± 0.2 (10) *↑ | 2.324423 ± 0.3 (9) **↑ |
| Ptgs2 | 4.27398 ± 0.32 (8) ****↑ | 3.89243 ± 0.2 (9) ****↑ | 4.05906 ± 0.14 (10) ****↑ | 3.86385 ± 0.46 (9) ****↑ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlost, J.; Kostrzewa, M.; Malek, N.; Starowicz, K. Molecular Understanding of the Activation of CB1 and Blockade of TRPV1 Receptors: Implications for Novel Treatment Strategies in Osteoarthritis. Int. J. Mol. Sci. 2018, 19, 342. https://doi.org/10.3390/ijms19020342
Mlost J, Kostrzewa M, Malek N, Starowicz K. Molecular Understanding of the Activation of CB1 and Blockade of TRPV1 Receptors: Implications for Novel Treatment Strategies in Osteoarthritis. International Journal of Molecular Sciences. 2018; 19(2):342. https://doi.org/10.3390/ijms19020342
Chicago/Turabian StyleMlost, Jakub, Magdalena Kostrzewa, Natalia Malek, and Katarzyna Starowicz. 2018. "Molecular Understanding of the Activation of CB1 and Blockade of TRPV1 Receptors: Implications for Novel Treatment Strategies in Osteoarthritis" International Journal of Molecular Sciences 19, no. 2: 342. https://doi.org/10.3390/ijms19020342
APA StyleMlost, J., Kostrzewa, M., Malek, N., & Starowicz, K. (2018). Molecular Understanding of the Activation of CB1 and Blockade of TRPV1 Receptors: Implications for Novel Treatment Strategies in Osteoarthritis. International Journal of Molecular Sciences, 19(2), 342. https://doi.org/10.3390/ijms19020342