High Throughput Chemical Screening Reveals Multiple Regulatory Proteins on FOXA1 in Breast Cancer Cell Lines

Abstract
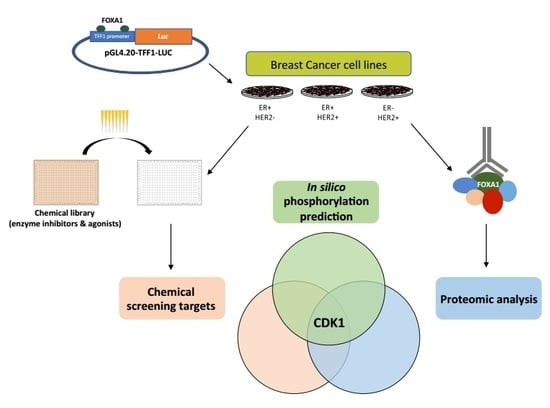
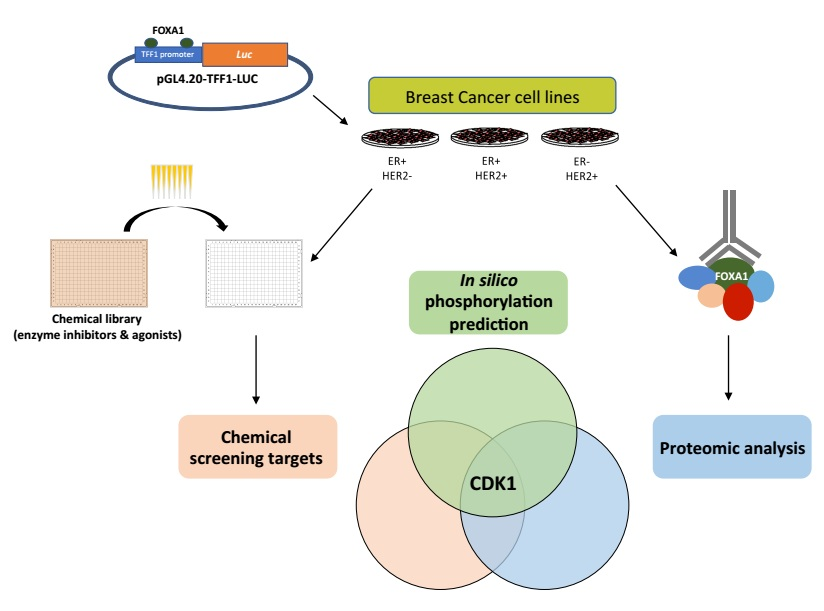{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
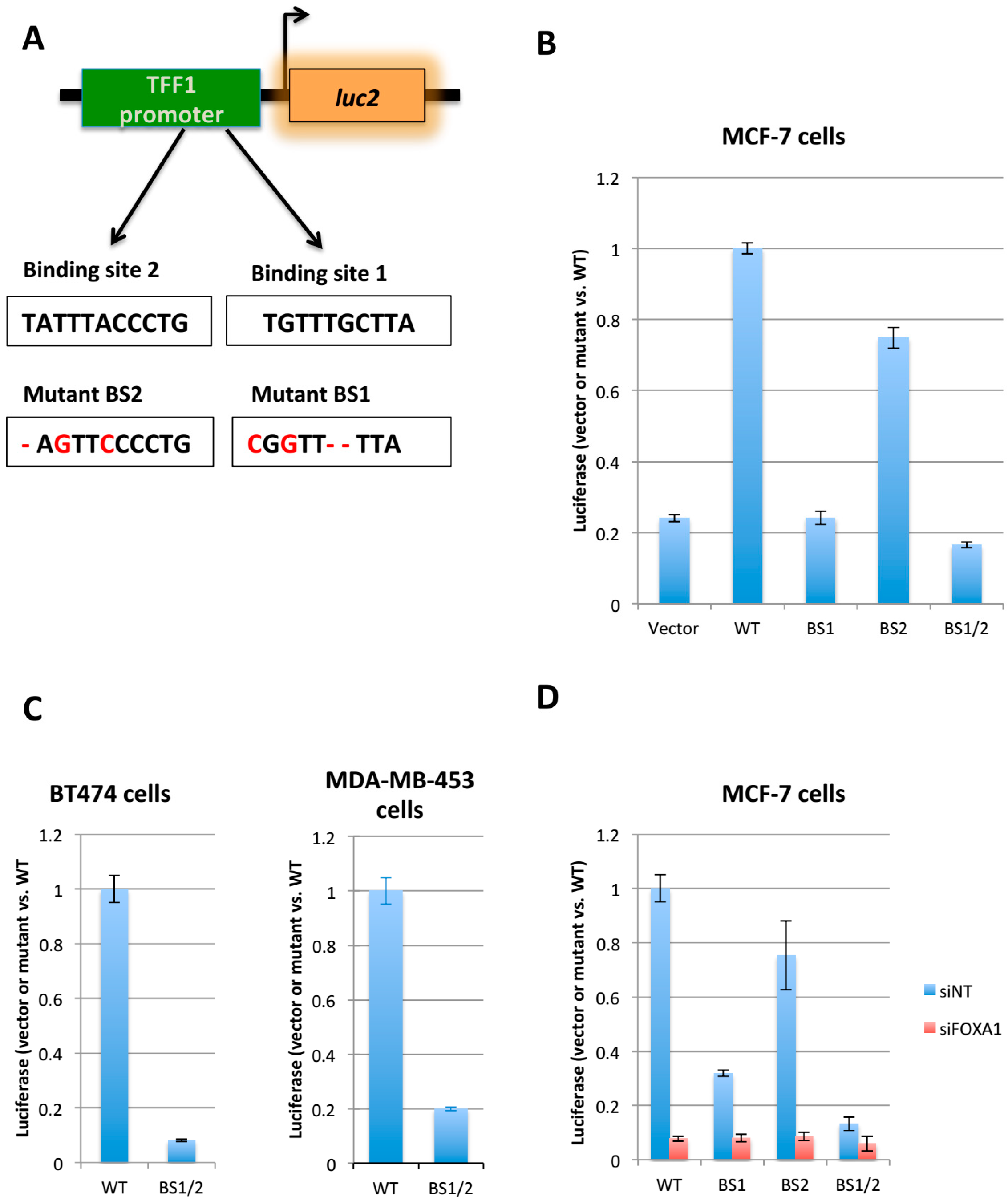
2.1. Generation of the FOXA1 Luciferase Reporter System
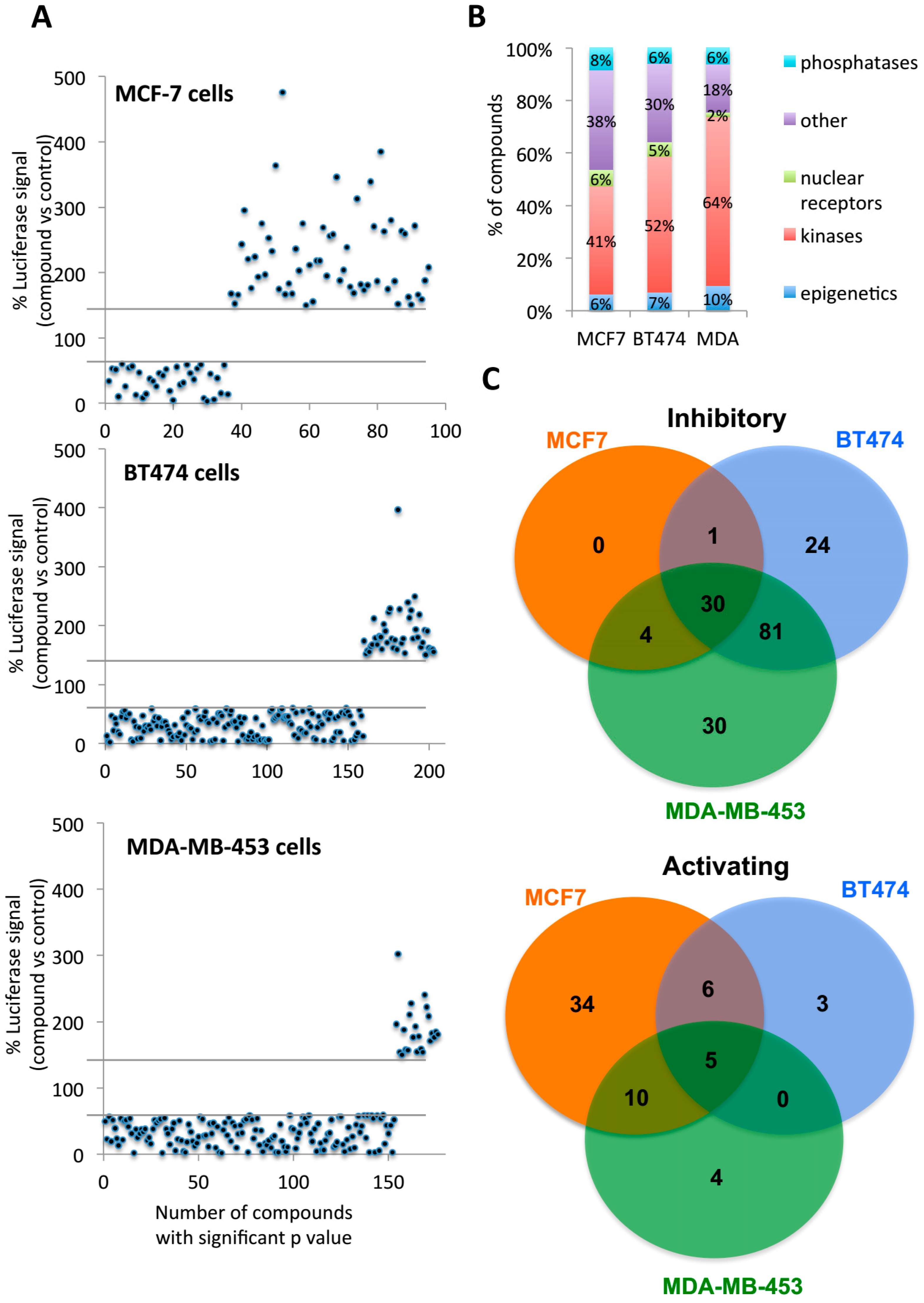
2.2. Multiple Targets Were Identified as Potential FOXA1 Regulators
2.3. Second Screening Narrowed down the Number of Compound Target Candidates
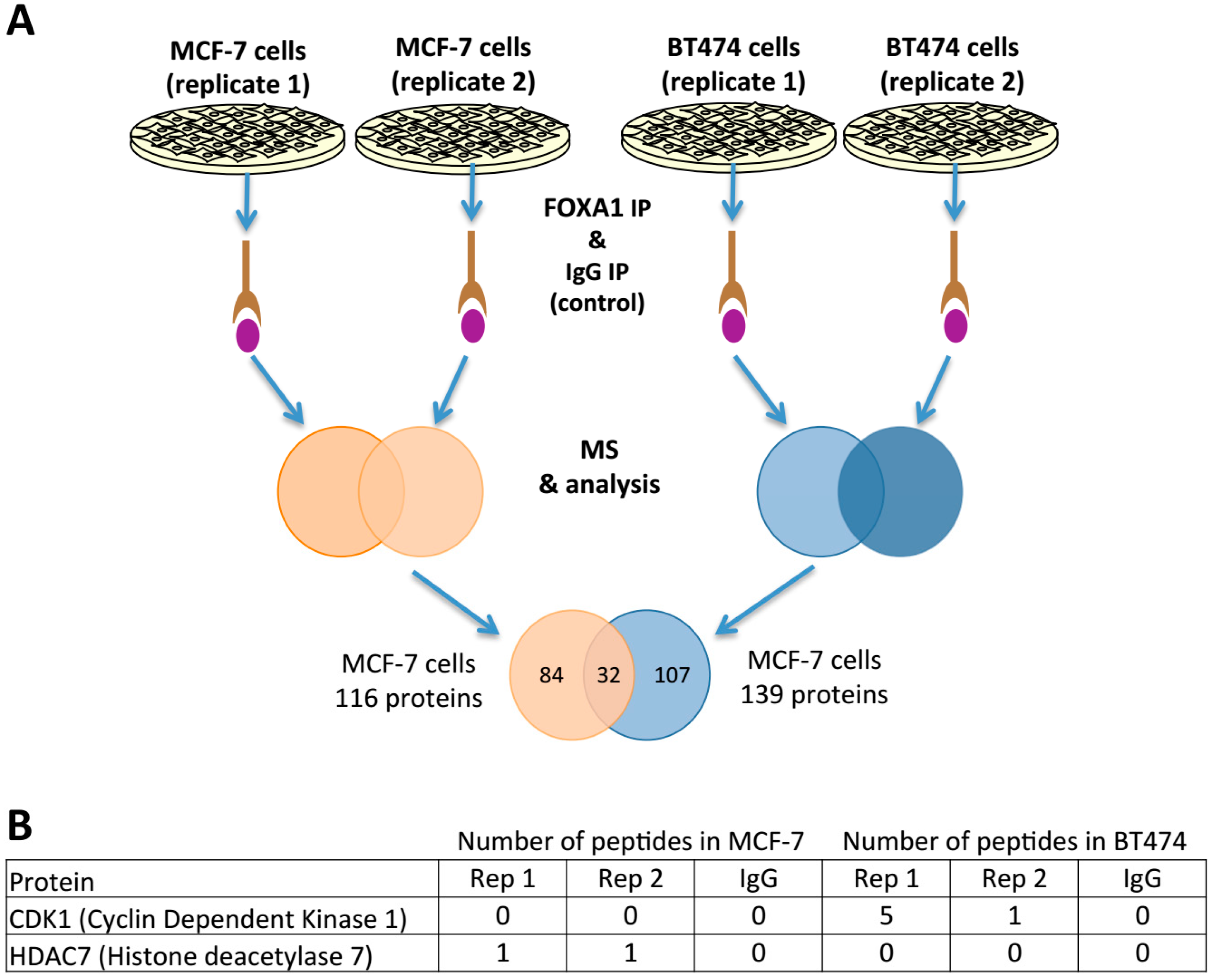
2.4. FOXA1 Pulldown and Proteomics Identify CDK1 as a Potential Direct Regulator
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construction of the Luciferase Reporter System
4.3. Transfection and Luciferase Assay
4.4. Chromatin Immunoprecipitation
4.5. High Throughput Chemical Screening and Analysis
4.6. FOXA1 Phosphorylation Sites Prediction
4.7. Immunoprecipitation
4.8. Protein Digestion
4.9. Desalting Digested Peptides
4.10. Data Processing and Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mirosevich, J.; Gao, N.; Matusik, R.J. Expression of Foxa transcription factors in the developing and adult murine prostate. Prostate 2005, 62, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, G.M.; Lozada, K.L.; Miedler, J.D.; Harburg, G.; Hewitt, S.C.; Mosley, J.D.; Godwin, A.K.; Korach, K.S.; Visvader, J.E.; Kaestner, K.H.; et al. FOXA1 is an essential determinant of ERα expression and mammary ductal morphogenesis. Development 2010, 137, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.A.; McPherson, C.E.; Bossard, P.; Stevens, K.; Cherian, S.; Shim, E.Y.; Clark, K.L.; Burley, S.K.; Zaret, K.S. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998, 17, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of Compacted Chromatin by Early Developmental Transcription Factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 Translates Epigenetic Signatures into Enhancer-Driven Lineage-Specific Transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef]
- Sérandour, A.A.; Avner, S.; Percevault, F.; Demay, F.; Bizot, M.; Lucchetti-Miganeh, C.; Barloy-Hubler, F.; Brown, M.; Lupien, M.; Métivier, R.; et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011, 21, 555–565. [Google Scholar] [CrossRef]
- Jozwik, K.M.; Chernukhin, I.; Serandour, A.A.; Nagarajan, S.; Carroll, J.S. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 2016, 17, 2715–2723. [Google Scholar] [CrossRef]
- Yang, Y.A.; Zhao, J.C.; Fong, K.W.; Kim, J.; Li, S.; Song, C.; Song, B.; Zheng, B.; He, C.; Yu, J. FOXA1 potentiates lineage-specific enhancer activation through modulating TET1 expression and function. Nucleic Acids Res. 2016, 44, 8153–8164. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Ross Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-Wide Mapping of Estrogen Receptor Binding Reveals Long-Range Regulation Requiring the Forkhead Protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Yu, J. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis. 2015, 2, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef]
- Laganière, J.; Deblois, G.; Lefebvre, C.; Bataille, A.R.; Robert, F.; Giguère, V. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc. Natl. Acad. Sci. USA 2005, 102, 11651–11656. [Google Scholar] [CrossRef]
- Wang, B.; Wang, M.; Li, A. Prediction of post-translational modification sites using multiple kernel support vector machine. PeerJ 2017, 5, e3261. [Google Scholar] [CrossRef] [PubMed]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Yue, P.; Gal, A.A.; Khuri, F.R.; Sun, S.-Y. Maintaining Glycogen Synthase Kinase-3 Activity Is Critical for mTOR Kinase Inhibitors to Inhibit Cancer Cell Growth. Cancer Res. 2014, 74, 2555–2568. [Google Scholar] [CrossRef]
- Thorne, C.A.; Wichaidit, C.; Coster, A.D.; Posner, B.A.; Wu, L.F.; Altschuler, S.J. GSK-3 modulates cellular responses to a broad spectrum of kinase inhibitors. Nat. Chem. Biol. 2015, 11, 58–63. [Google Scholar] [CrossRef]
- Landry, B.D.; Mapa, C.E.; Arsenault, H.E.; Poti, K.E.; Benanti, J.A. Regulation of a transcription factor network by Cdk1 coordinates late cell cycle gene expression. EMBO J. 2014, 33, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- Caravaca, J.M.; Donahue, G.; Becker, J.S.; He, X.; Vinson, C.; Zaret, K.S. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013, 27, 251–260. [Google Scholar] [CrossRef]
- Gilfillan, S.; Fiorito, E.; Hurtado, A. Functional genomic methods to study estrogen receptor activity. J. Mammary Gland Biol. Neoplasia 2012, 17, 147–153. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Singh, S.K.; Katika, M.R.; Lopez-Aviles, S.; Hurtado, A. High Throughput Chemical Screening Reveals Multiple Regulatory Proteins on FOXA1 in Breast Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 4123. https://doi.org/10.3390/ijms19124123
Wang S, Singh SK, Katika MR, Lopez-Aviles S, Hurtado A. High Throughput Chemical Screening Reveals Multiple Regulatory Proteins on FOXA1 in Breast Cancer Cell Lines. International Journal of Molecular Sciences. 2018; 19(12):4123. https://doi.org/10.3390/ijms19124123
Chicago/Turabian StyleWang, Shixiong, Sachin Kumar Singh, Madhumohan R. Katika, Sandra Lopez-Aviles, and Antoni Hurtado. 2018. "High Throughput Chemical Screening Reveals Multiple Regulatory Proteins on FOXA1 in Breast Cancer Cell Lines" International Journal of Molecular Sciences 19, no. 12: 4123. https://doi.org/10.3390/ijms19124123
APA StyleWang, S., Singh, S. K., Katika, M. R., Lopez-Aviles, S., & Hurtado, A. (2018). High Throughput Chemical Screening Reveals Multiple Regulatory Proteins on FOXA1 in Breast Cancer Cell Lines. International Journal of Molecular Sciences, 19(12), 4123. https://doi.org/10.3390/ijms19124123