Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
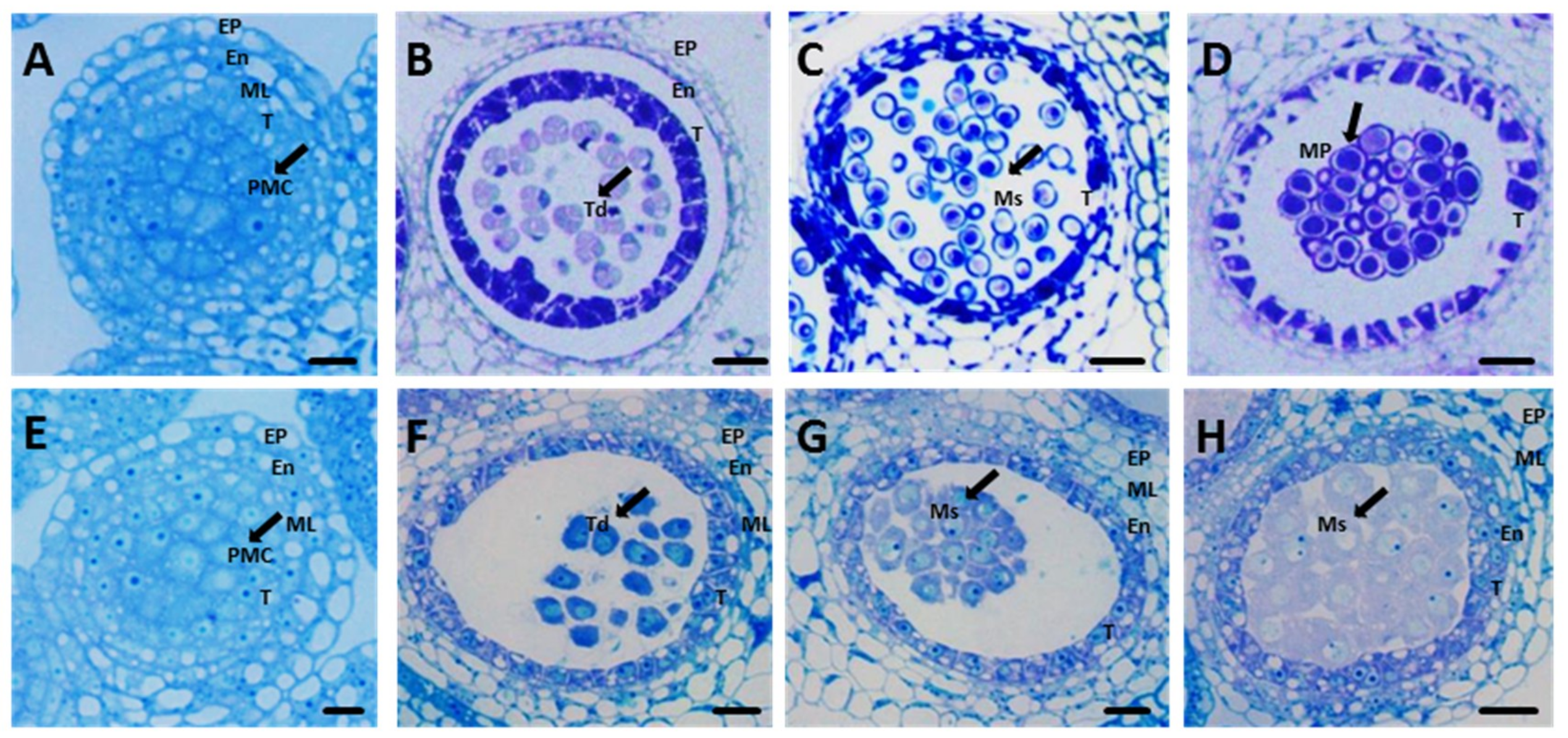
2.1. Phenotypic Characterization of CMS Lines and Maintainer Lines
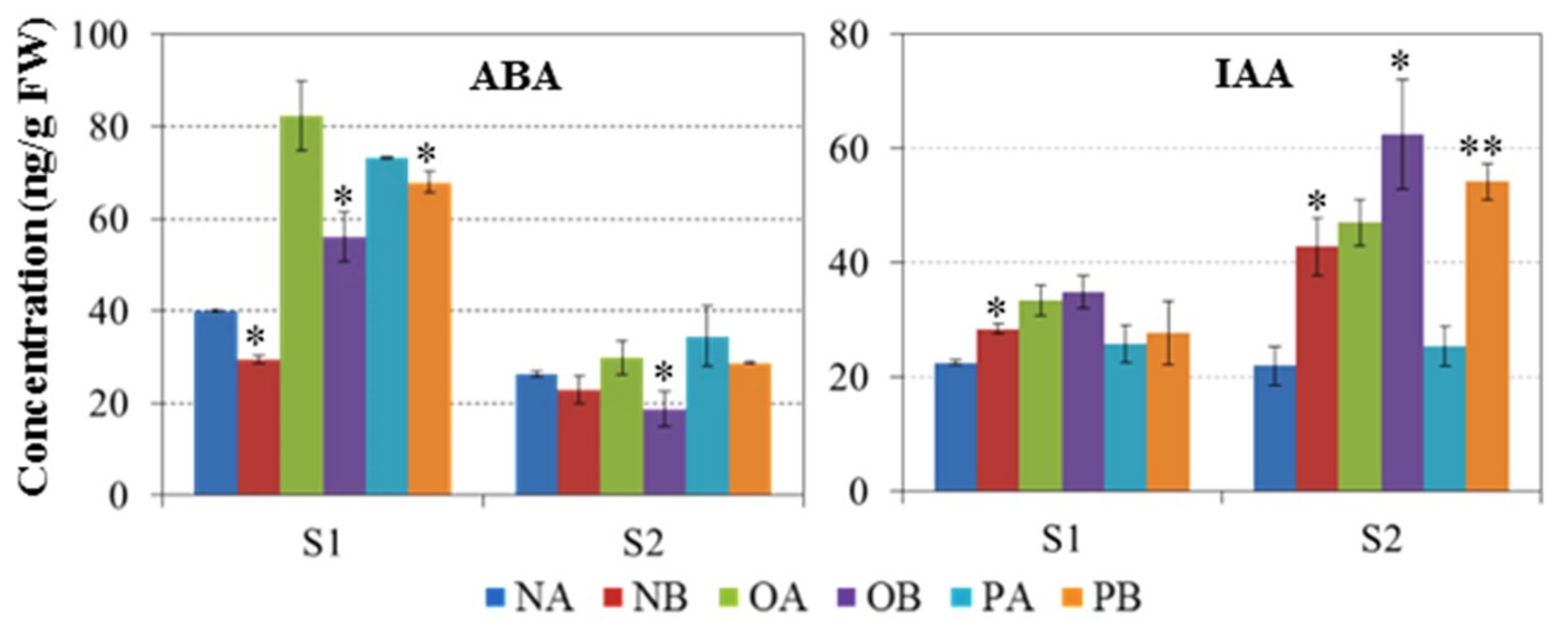
2.2. IAA and ABA Concentration in CMS and Maintainer Lines
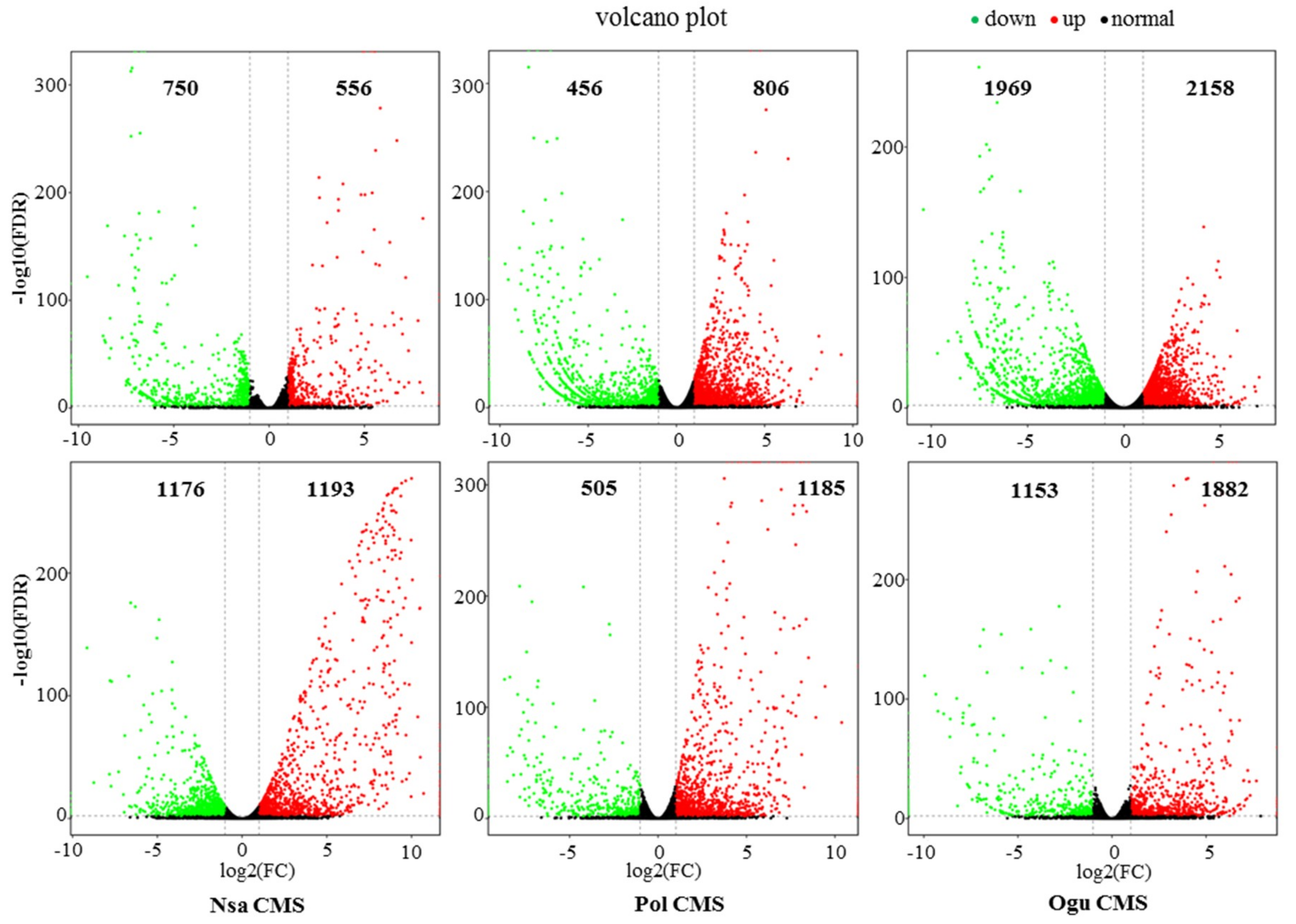
2.3. Differentially Expressed Genes in CMS and Maintainer Lines
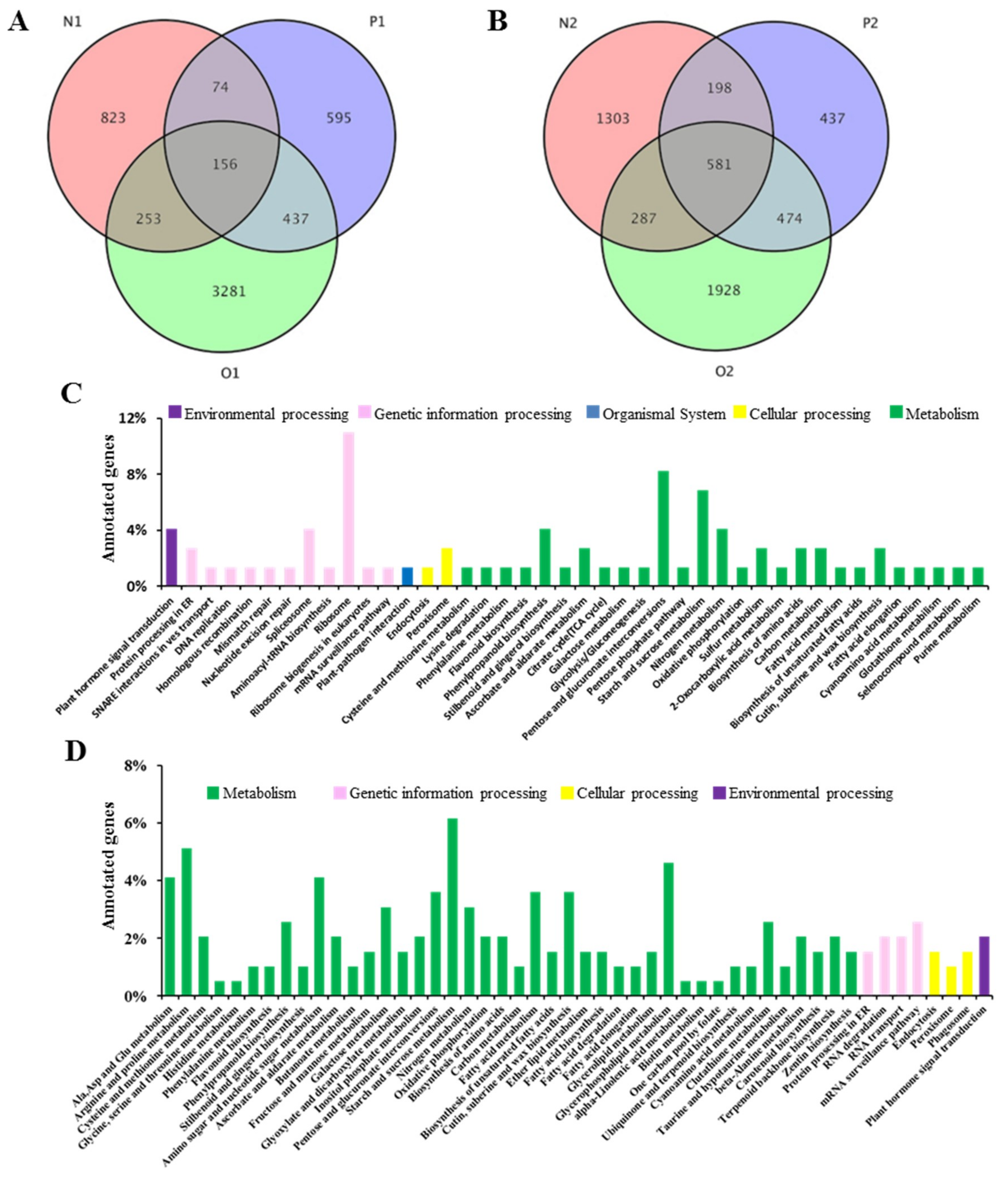
2.4. Gene Ontology and Classification of Three CMS Lines
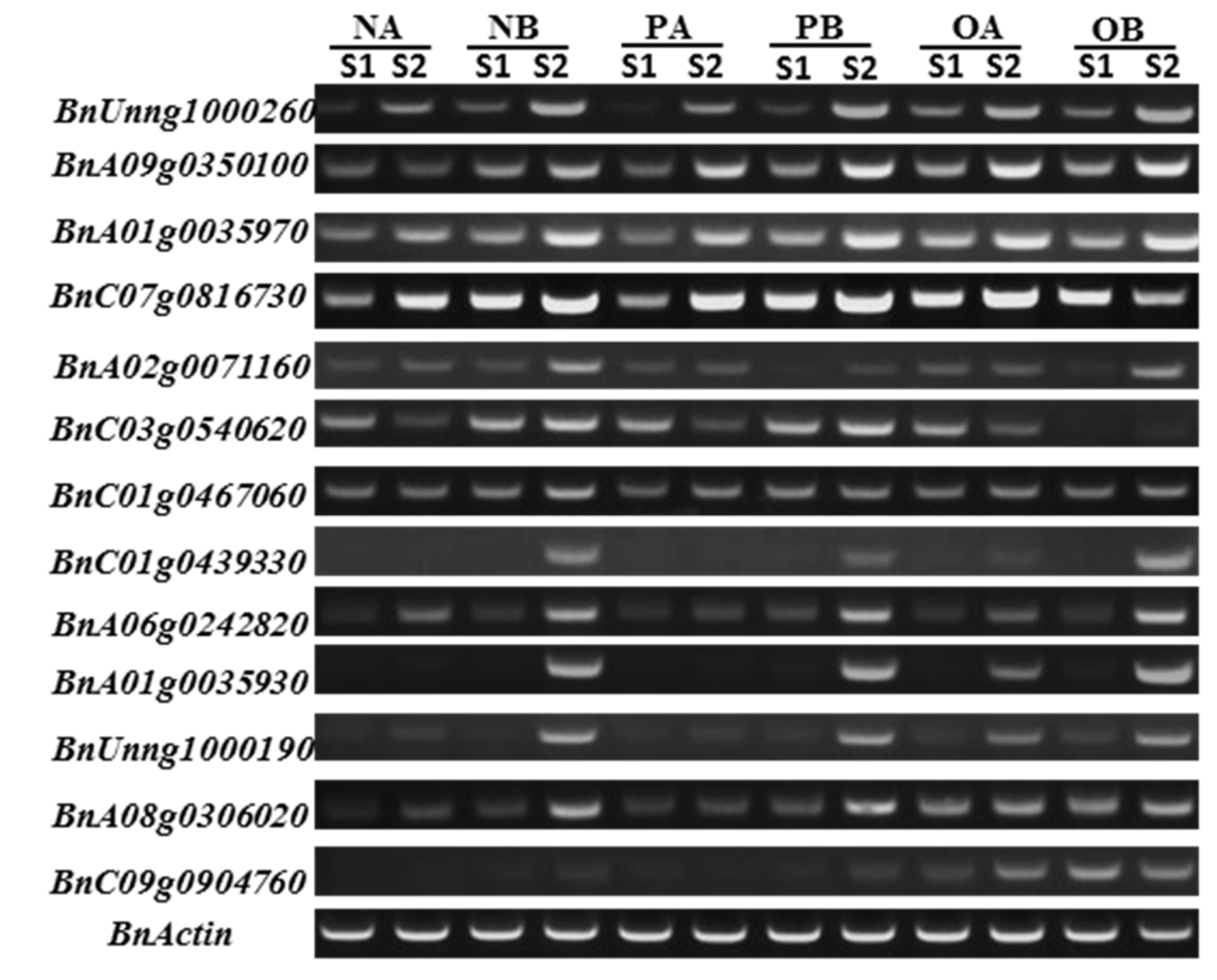
2.5. Verification of DEGs by RT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Morphology and Semi-Thin Sections
4.3. Phytohormone (ABA and IAA) Quantification
4.4. Illumina Sequencing and Analysis of DEGs
4.5. Semi-Quantitative (RT-PCR) Analysis of DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, H.Z. Review and future development of rapeseed industry in China. Chin. J. Oil Crop Sci. 2010, 2, 300–302. [Google Scholar]
- Fu, T.D. Breeding and utilization of rapeseed hybrid. Hubei Sci. Technol. 2000, 167–169. [Google Scholar]
- Allender, C.J.; King, G.J. Origins of the amphiploid species Brassica napus L. Investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 2010, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, H.; Bhat, S.R. Cytoplasmic male sterility in brassicaceae crops. Breed Sci. 2014, 64, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Shinada, T.; Kikuchi, Y.; Fujimoto, R.; Kishitani, S. An alloplasmic male-sterile line of Brassica oleracea harboring the mitochondria from Diplotaxis muralis expresses a novel chimeric open reading frame, orf72. Plant Cell Physiol. 2006, 47, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Shiga, T.; Baba, S. Cytoplasmic male sterility in oil seed rape. Brassica napus L., and its utilization to breeding. Japan J. Breed. 1973, 23, 187–197. [Google Scholar]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, 154–169. [Google Scholar] [CrossRef]
- Schnable, P.S.; Wise, R.P. The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 1998, 3, 175–180. [Google Scholar] [CrossRef]
- Horn, R.; Guptac, K.J.; Colombo, N. Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 2014, 19, 198–205. [Google Scholar] [CrossRef]
- Siedow, J.N.; Umbach, A.L. Plant mitochondrial electron transfer and molecular biology. Plant Cell 1995, 7, 821–831. [Google Scholar] [CrossRef]
- Logan, D.C. The mitochondrial compartment. J. Exp. Bot. 2006, 57, 1225–1243. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.D. Production and research of rapeseed in the People’s Republic of China. Eucarpia. Crucif. News 1981, 6, 6–7. [Google Scholar]
- Shen, J.X.; Wang, H.Z.; Fu, T.D.; Tian, B.M. Cytoplasmic male sterility with self-incompatibility, a novel approach to utilizing heterosis in rapeseed (Brassica napus L.). Euphytica 2008, 162, 109–115. [Google Scholar] [CrossRef]
- Thompson, K.F. Cytoplasmic male-sterility in oil-seed rape. Heredity 1972, 29, 253–257. [Google Scholar] [CrossRef]
- Hu, Q.; Andersen, S.; Dixelius, C.; Hansen, L. Production of fertile intergeneric somatic hybrids between brassica napus and sinapis arvensis for the enrichment of the rapeseed gene pool. Plant Cell Rep. 2002, 21, 147–152. [Google Scholar]
- Ogura, H. Studies on the new male-sterility in japanese radish, with special reference to the utilization of this sterility towerds the practical raising of hybrid seeds. Mém. Fac. Agric. Kagoshima Univ. 1967, 6, 1446–1459. [Google Scholar]
- Rawat, D.S.; Anand, I.J. Male sterility in indian mustard. Indian J. Genet. Plant Breed. 1979, 39, 412–414. [Google Scholar]
- Kemble, R.J.; Barsby, T.L. Use of protoplast fusion systems to study organelle genetics in a commercially important crop. Biochem. Cell Biol. 1988, 66, 665–676. [Google Scholar] [CrossRef]
- Burns, D.R.; Scarth, R.; McVetty, P.B.E. Temperature and genotypic effects on the expression of pol cytoplasmic male sterility in summer rape. Can. J. Plant Sci. 1991, 71, 655–661. [Google Scholar] [CrossRef]
- Zhang, J.K.; Zong, X.F.; Yu, G.D.; Li, J.N.; Zhang, W. Relationship between phytohormones and male sterility in thermo-photo-sensitive genic male sterile (TGMS) wheat. Euphytica 2006, 150, 241–248. [Google Scholar] [CrossRef]
- Zhou, Y.M.; Fu, T.D. Genetic improvement of rapeseed in China. In Proceedings of the 12th International Rapeseed Congress, Wuhan, China, 26–30 March 2007. [Google Scholar]
- Sawhney, V.K.; Shukla, A. Male sterility in flowering plants: Are plant growth substances involved? Am. J. Bot. 1994, 81, 1640–1647. [Google Scholar] [CrossRef]
- Tian, C.G.; Zhang, M.Y.; Duan, J. Preliminary study on the changes of phytohormones at different development stage in cytoplasmic male sterility line and its maintainer of rape. Sci. Agric. Sin. 1998, 31, 20–25. [Google Scholar]
- Wang, H.Z.; Wu, Z.D.; Han, Y. Relationships between endogenous hormone contents and cytoplasmic male sterility in sugarbeet. Sci. Agric. Sin. 2008, 41, 1134–1141. [Google Scholar]
- Wu, Z.M.; Hu, K.L.; Fu, J.Q.; Qiao, A.M. Relationships between cytoplasmic male sterility and endogenous hormone content of pepper bud. J. South China Agric. Univ. 2010, 31, 1–4. [Google Scholar]
- Dubas, E.; Janowiak, F.; Krzewska, M.; Hura, T.; Żur, I. Endogenous aba concentration and cytoplasmic membrane fluidity in microspores of oilseed rape (Brassica napus L.) genotypes differing in responsiveness to androgenesis induction. Plant Cell Rep. 2013, 32, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Sawhney, V.K. Abscisic acid: One of the factors affecting male sterility in Brassica napus. Physiol. Plant. 2010, 91, 522–528. [Google Scholar] [CrossRef]
- Guan, T.; Dang, Z.; Zhang, J. Studies on the changes of phytohormones during bud development stage in thermo-sensitivity genic male-sterile flax. Chin. J. Oil Crop Sci. 2007, 3, 248–253. [Google Scholar]
- Huang, S.; Zhou, X. Relationship between rice cytoplasmic male sterility and contents of GA (1+4) and IAA. Acta Agric. Bor-Sin. 1994, 9, 16–20. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.X.; Liu, Y.B.; Rong, W.H. RNA-Seq and its applications: A new technology for transcriptomics. Hereditas 2011, 33, 1191. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.; Fornara, F.; Vincent, C.; Andrés, F.; Nordström, K.; Göbel, U.; Knoll, D.; Schoof, H.; Coupland, G. Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering. Plant Cell. 2012, 24, 444–462. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Yang, Z.; Yi, B.; Wen, J.; Shen, J.; Tu, J.; Ma, C.; Fu, T. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genom. 2014, 15, 258. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Kang, J.H.; Zhao, M.; Kwon, J.K.; Choi, H.S.; Bae, J.H.; Lee, H.A.; Joung, Y.H.; Choi, D.; Kang, B.C. Tomato male sterile 1035 is essential for pollen development and meiosis in anthers. J. Exp. Bot. 2014, 65, 6693–6709. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, H.; Zheng, Y.; Ding, Y. Comparative expression profiling of mirnas between the cytoplasmic male sterile line meixianga and its maintainer line meixiangb during rice anther development. Planta 2015, 241, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Fu, F.; Liu, M.; Zhao, H.; Liu, C.; Li, J.; Tang, Z.; Xu, X.; Qiu, X.; Wang, R.; et al. Comparative transcriptome analysis of recessive male sterility (RGMS) in sterile and fertile Brassica napus lines. PLoS ONE 2015, 10, e0144118. [Google Scholar] [CrossRef]
- Du, K.; Liu, Q.; Wu, X.Y.; Jiang, J.J.; Wu, J.; Fang, Y.J.; Li, A.M.; Wang, Y. Morphological structure and transcriptome comparison of the cytoplasmic male sterility line in Brassica napus (SaNa-1A) derived from somatic hybridization and its maintainer line SaNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef]
- Leino, M.; Teixeira, R.; Landgren, M.; Glimelius, K. Brassica napus lines with rearranged arabidopsis mitochondria display CMS and a range of developmental aberrations. Theor. Appl. Genet. 2003, 106, 1156–1163. [Google Scholar] [CrossRef]
- Mackenzie, S. Male sterility and hybrid seed production. Plant Biotechnol. Agric. 2012, 185–194. [Google Scholar]
- Datta, R.; Chamusco, K.C.; Chourey, P.S. Starch biosynthesis during pollen maturation is associated with altered patterns of gene expression in maize. Plant Physiol. 2002, 130, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- González-Melendi, P.; Uyttewaal, M.; Morcillo, C.N.; Hernández Mora, J.R.; Fajardo, S.; Budar, F.; Lucas, M.M. A light and electron microscopy analysis of the events leading to male sterility in Ogu-INRA CMS of rapeseed (Brassica napus). J. Exp. Bot. 2008, 59, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Huai, Y.; Zhang, M.F. Mitochondrial atpa gene is altered in a new orf220-type cytoplasmic male-sterile line of stem mustard (Brassica juncea). Mol. Biol. Rep. 2009, 36, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.D.; Dai, L.F.; Wang, S.B.; Wolukau, J.; Jahn, M.; Chen, J.F. Male gamete development and early tapetal degeneration in cytoplasmic male sterile pepper investigated by meiotic, anatomical and ultrastructural analyses. Plant Breed. 2006, 125, 395–399. [Google Scholar] [CrossRef]
- Li, N.; Zhang, D.S.; Liu, H.S.; Yin, C.S.; Li, X.X.; Liang, W.Q.; Yuan, Z.; Xu, B.; Chu, H.W.; Wang, J. The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef]
- Minocha, S.C. The role of auxin and abscisic acid in the induction of cell division in jerusalem artichoke tuber tissue cultured in vitro. Z. Pflanzenphysiol. 1979, 92, 431–441. [Google Scholar] [CrossRef]
- Rook, F.; Hadingham, S.A.; Li, Y.; Bevan, M.W. Sugar and aba response pathways and the control of gene expression. Plant Cell Environ. 2006, 29, 426–434. [Google Scholar] [CrossRef]
- Ji, X.; Dong, B.; Shiran, B.; Talbot, M.J.; Edlington, J.E.; Hughes, T.; White, R.G.; Gubler, F.; Dolferus, R. Control of abscisic acid catabolism and abscisic acid homeostasis is important for reproductive stage stress tolerance in cereals. Plant Physiol. 2011, 156, 647–662. [Google Scholar] [CrossRef]
- De Storme, N.; Geelen, D. The impact of environmental stress on male reproductive development in plants: Biological processes and molecular mechanisms. Plant Cell Environ. 2014, 37, 1–18. [Google Scholar]
- Duca, M. Genetic-phytohormonal interactions in male fertility and male sterility phenotype expression in sunflower (Helianthus annuus L.)/interacciones genético-fitohormonales en la expresión fenotípica de la androfertilidad y androesterilidad en girasol (helianthus annuus l.)/interactions génétiques phytohormonales dans l’expression phénotypique d’un male fertile et male sterile du tournesol (helianthus annuus L.). Helia 2008, 31, 27–38. [Google Scholar]
- Singh, S.; Sawhney, V. Abscisic acid in a male sterile tomato mutant and its regulation by low temperature. J. Exp. Bot. 1998, 49, 199–203. [Google Scholar] [CrossRef]
- Yang, J.H.; Zhang, M.F.; Yu, J.Q. Mitochondrial nad2 gene is co-transcripted with CMS-associated orfB gene in cytoplasmic male-sterile stem mustard (Brassica juncea). Mol. Biol. Rep. 2009, 36, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Heng, S.; Liu, S.; Xia, C.; Tang, H.; Xie, F.; Fu, T.; Wan, Z. Morphological and genetic characterization of a new cytoplasmic male sterility system (oxa CMS) in stem mustard (Brassica juncea). Theor. Appl. Genet. 2018, 131, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Sun, C.; Li, H.; Hu, S.; Lei, L.; Kang, J. Integrated analysis of transcriptome and proteome changes related to the Ogura cytoplasmic male sterility in cabbage. PLoS ONE 2018, 13, e0193462. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ding, X.; Wang, X.; He, T.; Zhang, H.; Yang, L.; Wang, T.; Chen, L.; Gai, J.; Yang, S. Genome-wide comparative analysis of DNA methylation between soybean cytoplasmic male-sterile line NJCMS5A and its maintainer NJCMS5B. BMC Genom. 2017, 18, 596. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, H.; Wang, W.; Liu, J.; Hao, M.; Mei, D.; Zhou, R.; Fu, L.; Hu, Q. Identification of BnaYUCCA6 as a candidate gene for branch angle in Brassica napus by QTL-seq. Sci. Rep. 2016, 6, 38493. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Hao, M.; Wang, W.; Mei, D.; Wells, R.; Liu, J.; Wang, H.; Sang, S.; Tang, M.; Zhou, R.; et al. Integrative RNA- and miRNA-Profile Analysis Reveals a Likely Role of BR and Auxin Signaling in Branch Angle Regulation of B. napus. Int. J. Mol. Sci. 2017, 18, 887. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, B.; Hao, M.; Mei, D.; Zaman, Q.U.; Sang, S.; Wang, H.; Wang, W.; Fu, L.; Cheng, H.; Hu, Q. Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus . Int. J. Mol. Sci. 2018, 19, 4022. https://doi.org/10.3390/ijms19124022
Ding B, Hao M, Mei D, Zaman QU, Sang S, Wang H, Wang W, Fu L, Cheng H, Hu Q. Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus . International Journal of Molecular Sciences. 2018; 19(12):4022. https://doi.org/10.3390/ijms19124022
Chicago/Turabian StyleDing, Bingli, Mengyu Hao, Desheng Mei, Qamar U Zaman, Shifei Sang, Hui Wang, Wenxiang Wang, Li Fu, Hongtao Cheng, and Qiong Hu. 2018. "Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus " International Journal of Molecular Sciences 19, no. 12: 4022. https://doi.org/10.3390/ijms19124022
APA StyleDing, B., Hao, M., Mei, D., Zaman, Q. U., Sang, S., Wang, H., Wang, W., Fu, L., Cheng, H., & Hu, Q. (2018). Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus . International Journal of Molecular Sciences, 19(12), 4022. https://doi.org/10.3390/ijms19124022