Glucocorticoid Receptor Binding Inhibits an Intronic IL33 Enhancer and is Disrupted by rs4742170 (T) Allele Associated with Specific Wheezing Phenotype in Early Childhood


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
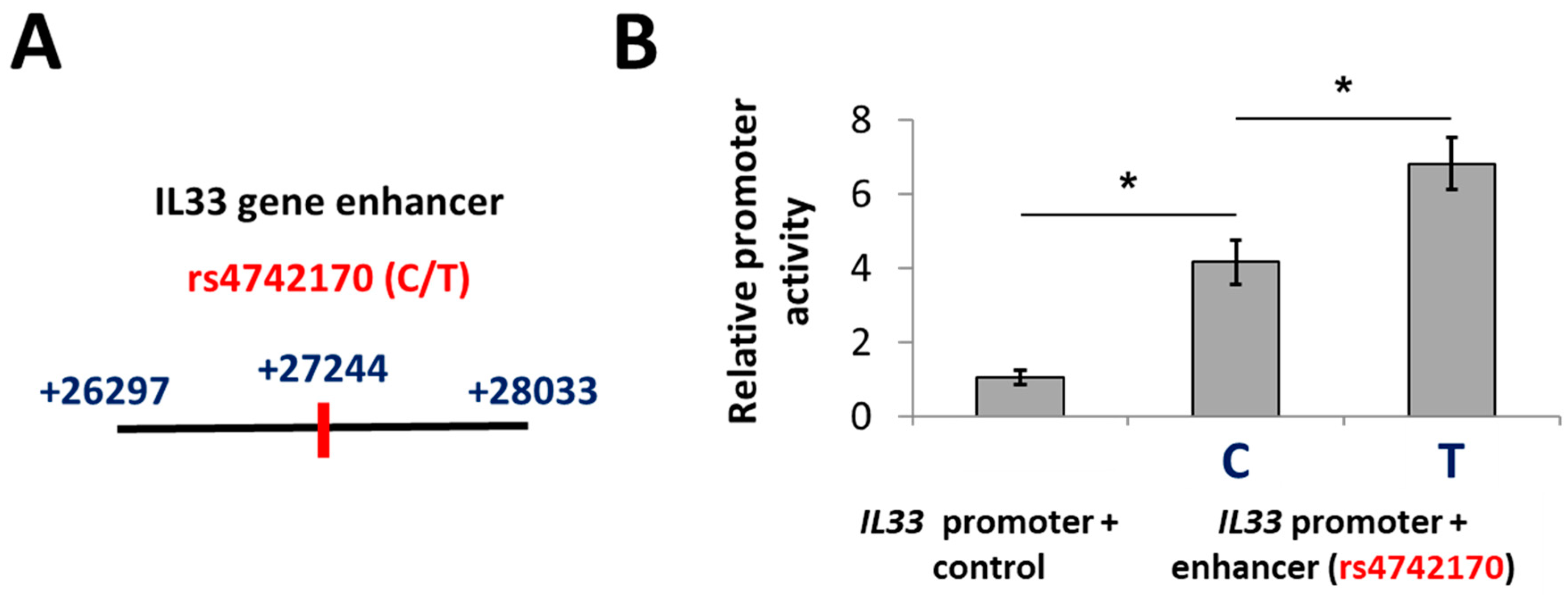
2.1. A Putative Enhancer Including rs4742170 Stimulates IL33 Promoter Activity
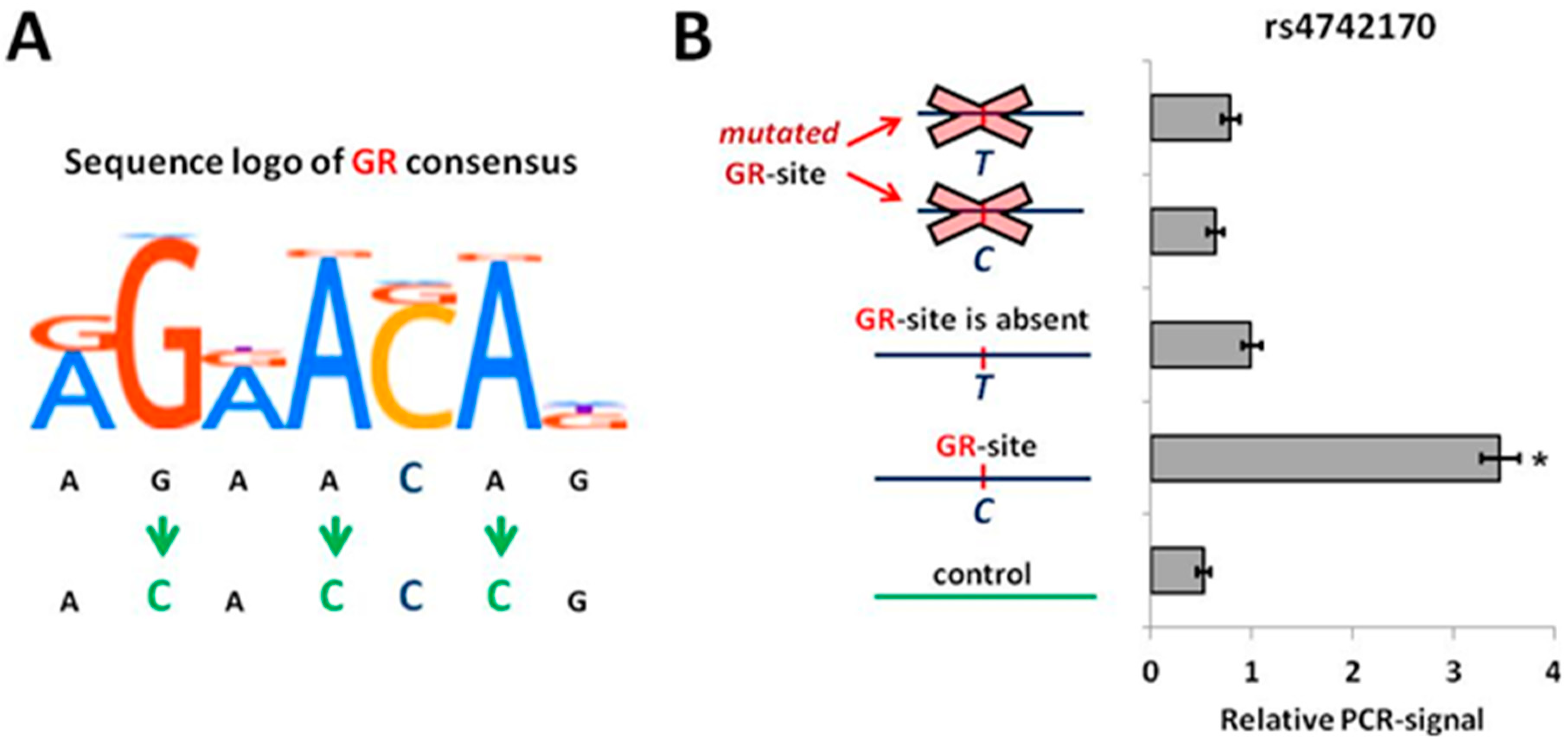
2.2. The rs4742170 (T) Allele in IL33 Enhancer Disrupts GR-Binding Site
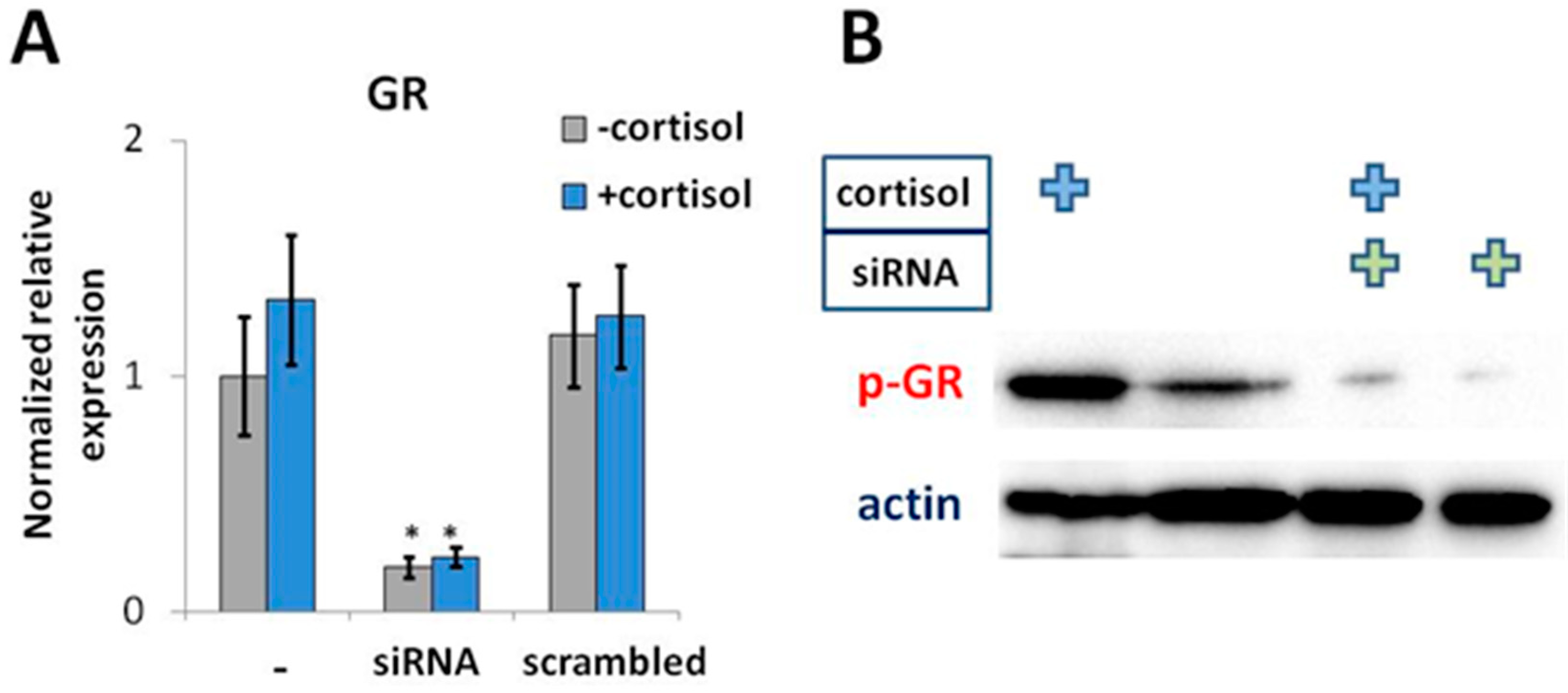
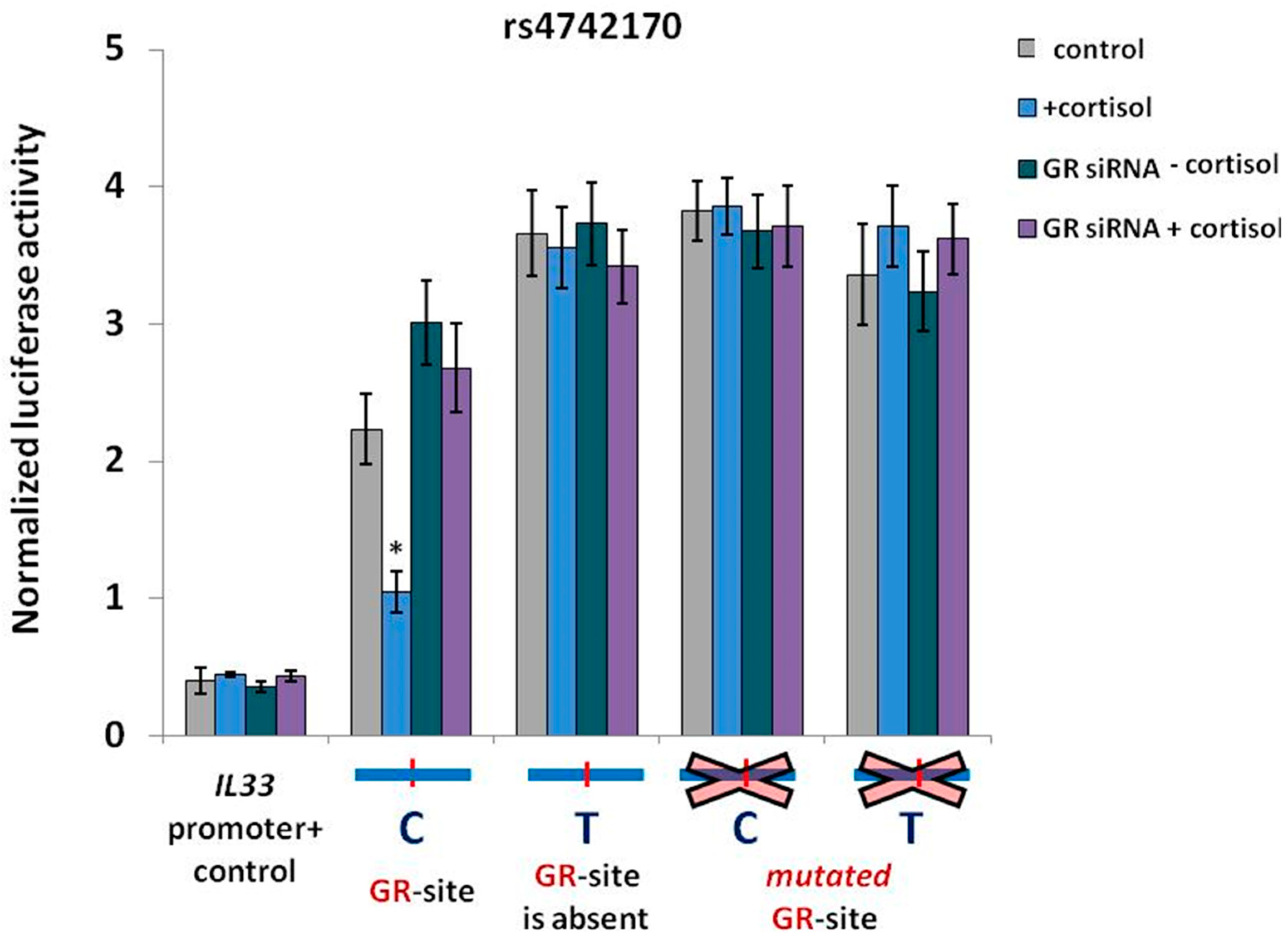
2.3. GR Activation is Associated with a Decrease in IL33 Promoter Activity
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Ethical Approval
4.3. Luciferase Reporter Constructs
4.4. Pull-Down Assay
4.5. GR Knockdown Using siRNA
4.6. Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [PubMed]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: In situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Weiss, T.M.; Niebuhr, M.; Pan, B.; Appleton, B.A.; Wiesmann, C.; Bazan, J.F.; Fairbrother, W.J. Structure of IL-33 and Its Interaction with the ST2 and IL-1RAcP Receptors-Insight into Heterotrimeric IL-1 Signaling Complexes. Structure 2009, 17, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Smith, D.E. IL-33: A tissue derived cytokine pathway involved in allergic inflammation and asthma. Clin. Exp. Allergy 2009, 40, 200–208. [Google Scholar] [CrossRef]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Matsuki, A.; Takatori, H.; Makita, S.; Yokota, M.; Tamachi, T.; Suto, A.; Suzuki, K.; Hirose, K.; Nakajima, H. T-bet inhibits innate lymphoid cell–mediated eosinophilic airway inflammation by suppressing IL-9 production. J. Allergy Clin. Immunol. 2016, 139, 1355–1367.e6. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Arron, J.R.; Koth, L.L.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Kewin, P.; Murphy, G.; Russo, R.C.; Stolarski, B.; Garcia, C.C.; Komai-Koma, M.; Pitman, N.; Li, Y.; McKenzie, A.N.J.; et al. IL-33 Induces Antigen-Specific IL-5+ T Cells and Promotes Allergic-Induced Airway Inflammation Independent of IL-4. J. Immunol. 2008, 181, 4780–4790. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Huang, P.; Zhang, Q.; Yao, X.; Wang, J.; Lv, Z.; An, Y.; Corrigan, C.J.; Huang, K.; et al. Distinct sustained structural and functional effects of interleukin-33 and interleukin-25 on the airways in a murine asthma surrogate. Immunology 2015, 145, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int. Immunol. 2008, 20, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Préfontaine, D.; Nadigel, J.; Chouiali, F.; Audusseau, S.; Semlali, A.; Chakir, J.; Martin, J.G.; Hamid, Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 2010, 125, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Hamzaoui, A.; Berraies, A.; Kaabachi, W.; Haifa, M.; Ammar, J.; Kamel, H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J. Asthma 2013, 50, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.W.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2018, 141, 257–268. [Google Scholar] [CrossRef]
- Kaur, D.; Gomez, E.; Doe, C.; Berair, R.; Woodman, L.; Saunders, R.; Hollins, F.; Rose, F.R.; Amrani, Y.; May, R.; et al. IL-33 drives airway hyper-responsiveness through IL-13-mediated mast cell: Airway smooth muscle crosstalk. Allergy Eur. J. Allergy Clin. Immunol. 2015, 70, 556–567. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Bjornsdottir, U.S.; Halapi, E.; Helgadottir, A.; Sulem, P.; Jonsdottir, G.M.; Thorleifsson, G.; Helgadottir, H.; Steinthorsdottir, V.; Stefansson, H.; et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 2009, 41, 342–347. [Google Scholar] [CrossRef]
- Moffatt, M.; Gut, I.; Demenais, F.; Strachan, D.; Bouzigon, E.; Heath, S. A Large-Scale, Consortium-Based Genomewide Association Study of Asthma. N. Engl. J. Med. 2010, 363, 525–527. [Google Scholar] [CrossRef]
- Torgerson, D.G.; Ampleford, E.J.; Chiu, G.Y.; Gauderman, W.J.; Gignoux, C.R.; Graves, P.E.; Himes, B.E.; Levin, A.M.; Mathias, R.A.; Hancock, D.B.; et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 2011, 43, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Bonnelykke, K.; Sleiman, P.; Nielsen, K.; Kreiner-Moller, E.; Mercader, J.M.; Belgrave, D.; Den Dekker, H.T.; Husby, A.; Sevelsted, A.; Faura-Tellez, G.; et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 2014, 46, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Savenije, O.E.; Mahachie John, J.M.; Granell, R.; Kerkhof, M.; Dijk, F.N.; De Jongste, J.C.; Smit, H.A.; Brunekreef, B.; Postma, D.S.; Van Steen, K.; et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J. Allergy Clin. Immunol. 2014, 134, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Gorbacheva, A.; Korneev, K.; Kuprash, D.; Mitkin, N. The Risk G Allele of the Single-Nucleotide Polymorphism rs928413 Creates a CREB1-Binding Site That Activates IL33 Promoter in Lung Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 2911. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.S.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. ENCODE Data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2013, 41, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Vorontsov, I.E.; Kulakovskiy, I.V.; Khimulya, G.; Nikolaeva, D.D.; Makeev, V.J. PERFECTOS-APE: Predicting regulatory functional effect of SNPs by approximate P-value estimation. In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms, Lisbon, Portugal, 12–15 January 2015; pp. 102–108. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, S.; Anderson, D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 2000, 25, 331–343. [Google Scholar] [CrossRef]
- Takebayashi, H.; Yoshida, S.; Sugimori, M.; Kosako, H.; Kominami, R.; Nakafuku, M.; Nabeshima, Y.I. Dynamic expression of basic helix-loop-helix Olig family members: Implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech. Dev. 2000, 99, 143–148. [Google Scholar] [CrossRef]
- Mellentin, J.D.; Smith, S.D.; Cleary, M.L. Lyl-1, a novel gene altered by chromosomal translocation in T cell leukemia, codes for a protein with a helix-loop-helix DNA binding motif. Cell 1989, 58, 77–83. [Google Scholar] [CrossRef]
- Souroullas, G.P.; Salmon, J.M.; Sablitzky, F.; Curtis, D.J.; Goodell, M.A. Adult Hematopoietic Stem and Progenitor Cells Require Either Lyl1 or Scl for Survival. Cell Stem Cell 2009, 4, 180–186. [Google Scholar] [CrossRef]
- Brinkmann, A.O.; Trapman, J. Genetic analysis of androgen receptors in development and disease. Adv. Pharmacol. 2000, 47, 317–341. [Google Scholar]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Le Cam, L.; Lacroix, M.; Ciemerych, M.A.; Sardet, C.; Sicinski, P. The E4F Protein Is Required for Mitotic Progression during Embryonic Cell Cycles. Mol. Cell. Biol. 2004, 24, 6467–6475. [Google Scholar] [CrossRef] [PubMed]
- Chagraoui, J.; Niessen, S.; Lessard, J.; Girard, S.; Coulombe, P.; Meloche, S.; Sauvageau, G. p120E4F-1: A novel candidate factor for mediating Bmi-1 function in hematopoietic stem cells. Blood 2004, 104, 109A. [Google Scholar] [CrossRef]
- Hatchi, E.; Rodier, G.; Lacroix, M.; Caramel, J.; Kirsh, O.; Jacquet, C.; Schrepfer, E.; Lagarrigue, S.; Linares, L.K.; Lledo, G.; et al. E4F1 deficiency results in oxidative stress-mediated cell death of leukemic cells. J. Exp. Med. 2011, 208, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- Chandler, V.L.; Maler, B.A.; Yamamoto, K.R. DNA sequences bound specifically by glucocorticoid receptor in vitro render a heterologous promoter hormone responsive in vivo. Cell 1983, 33, 489–499. [Google Scholar] [CrossRef]
- Adcock, I.M.; Gilbey, T.; Gelder, C.M.; Chung, K.F.; Barnes, P.J. Glucocorticoid receptor localization in normal and asthmatic lung. Am. J. Respir. Crit. Care Med. 1996, 154, 771–782. [Google Scholar] [CrossRef]
- Funder, J.W. Glucocorticoid and Mineralocorticoid Receptors: Biology and Clinical Relevance. Annu. Rev. Med. 1997, 48, 231–240. [Google Scholar] [CrossRef]
- Barnes, P.J. New drugs for asthma. Semin. Respir. Crit. Care Med. 2012, 33, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Adcock, I. Pharmacology of airway inflammation in asthma and COPD. Pulm. Pharmacol. Ther. 2003, 16, 247–277. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Leigh, R.; Giembycz, M.A. Pharmacological strategies for improving the efficacy and therapeutic ratio of glucocorticoids in inflammatory lung diseases. Pharmacol. Ther. 2010, 125, 286–327. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M. Widespread Negative Response Elements Mediate Direct Repression by Agonist- Liganded Glucocorticoid Receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [PubMed]
- Uhlenhaut, N.H.; Barish, G.D.; Yu, R.T.; Downes, M.; Karunasiri, M.; Liddle, C.; Schwalie, P.; Hübner, N.; Evans, R.M. Insights into Negative Regulation by the Glucocorticoid Receptor from Genome-wide Profiling of Inflammatory Cistromes. Mol. Cell 2013, 49, 158–171. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Haegeman, G. Minireview: Latest Perspectives on Antiinflammatory Actions of Glucocorticoids. Mol. Endocrinol. 2009, 23, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 33, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Beato, M.; Herrlich, P.; Schütz, G. Steroid hormone receptors: Many Actors in search of a plot. Cell 1995, 83, 851–857. [Google Scholar] [CrossRef]
- Barnes, P.J. Glucocorticosteroids. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2016; Volume 237, pp. 93–115. [Google Scholar]
- Saglani, S.; Lui, S.; Ullmann, N.; Campbell, G.A.; Sherburn, R.T.; Mathie, S.A.; Denney, L.; Bossley, C.J.; Oates, T.; Walker, S.A.; et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J. Allergy Clin. Immunol. 2013, 132, 676–685. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Lv, Z.; Li, Y.; Chen, Y.; Huang, K.; Corrigan, C.J.; Ying, S. Elevated Expression of IL-33 and TSLP in the Airways of Human Asthmatics In Vivo: A Potential Biomarker of Severe Refractory Disease. J. Immunol. 2018, 200, 2253–2262. [Google Scholar] [CrossRef]
- Ding, W.; Zou, G.; Zhang, W.; Lai, X.; Chen, H.; Xiong, L. Interleukin-33: Its Emerging Role in Allergic Diseases. Molecules 2018, 23, 1665. [Google Scholar] [CrossRef]
- Nabe, T.; Wakamori, H.; Yano, C.; Nishiguchi, A.; Yuasa, R.; Kido, H.; Tomiyama, Y.; Tomoda, A.; Kida, H.; Takiguchi, A.; et al. Production of interleukin (IL)-33 in the lungs during multiple antigen challenge-induced airway inflammation in mice, and its modulation by a glucocorticoid. Eur. J. Pharmacol. 2015, 757, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.D.; O’Byrne, P.M. Biologics and the lung: TSLP and other epithelial cell-derived cytokines in asthma. Pharmacol. Ther. 2016, 169, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Wang, L. In vitro human cell line models to predict clinical response to anticancer drugs. Pharmacogenomics 2015, 16, 273–285. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, W.; Liyanarachchi, S.; Srinivas, M.; Wang, Y.; Akagi, K.; Wang, Y.; Wu, D.; Wang, Q.; Jin, V.; et al. Multiple functional variants in long-range enhancer elements contribute to the risk of SNP rs965513 in thyroid cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 6128–6133. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.F.; Peterson, C.L.; Smale, S.T. Confirming the functional importance of a protein-DNA interaction. Cold Spring Harb. Protoc. 2012, 7, 733–757. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Mitkin, N.A.; Hook, C.D.; Schwartz, A.M.; Biswas, S.; Kochetkov, D.V.; Muratova, A.M.; Afanasyeva, M.A.; Kravchenko, J.E.; Bhattacharyya, A.; Kuprash, D.V. p53-dependent expression of CXCR5 chemokine receptor in MCF-7 breast cancer cells. Sci. Rep. 2015, 5, 9330. [Google Scholar] [CrossRef]
- Mitkin, N.A.; Muratova, A.M.; Schwartz, A.M.; Kuprash, D.V. The A allele of the Single-Nucleotide Polymorphism rs630923 creates a Binding site for MEF2C resulting in reduced CXCR5 Promoter activity in B-cell lymphoblastic cell lines. Front. Immunol. 2016, 7, 515. [Google Scholar] [CrossRef]
- Mitkin, N.A.; Muratova, A.M.; Korneev, K.V.; Pavshintsev, V.V.; Rumyantsev, K.A.; Vagida, M.S.; Uvarova, A.N.; Afanasyeva, M.A.; Schwartz, A.M.; Kuprash, D.V. Protective C allele of the single-nucleotide polymorphism rs1335532 is associated with strong binding of Ascl2 transcription factor and elevated CD58 expression in B-cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3211–3220. [Google Scholar] [CrossRef]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorbacheva, A.M.; Kuprash, D.V.; Mitkin, N.A. Glucocorticoid Receptor Binding Inhibits an Intronic IL33 Enhancer and is Disrupted by rs4742170 (T) Allele Associated with Specific Wheezing Phenotype in Early Childhood. Int. J. Mol. Sci. 2018, 19, 3956. https://doi.org/10.3390/ijms19123956
Gorbacheva AM, Kuprash DV, Mitkin NA. Glucocorticoid Receptor Binding Inhibits an Intronic IL33 Enhancer and is Disrupted by rs4742170 (T) Allele Associated with Specific Wheezing Phenotype in Early Childhood. International Journal of Molecular Sciences. 2018; 19(12):3956. https://doi.org/10.3390/ijms19123956
Chicago/Turabian StyleGorbacheva, Alisa M., Dmitry V. Kuprash, and Nikita A. Mitkin. 2018. "Glucocorticoid Receptor Binding Inhibits an Intronic IL33 Enhancer and is Disrupted by rs4742170 (T) Allele Associated with Specific Wheezing Phenotype in Early Childhood" International Journal of Molecular Sciences 19, no. 12: 3956. https://doi.org/10.3390/ijms19123956
APA StyleGorbacheva, A. M., Kuprash, D. V., & Mitkin, N. A. (2018). Glucocorticoid Receptor Binding Inhibits an Intronic IL33 Enhancer and is Disrupted by rs4742170 (T) Allele Associated with Specific Wheezing Phenotype in Early Childhood. International Journal of Molecular Sciences, 19(12), 3956. https://doi.org/10.3390/ijms19123956