Calcium Signaling in Cholangiocytes: Methods, Mechanisms, and Effects


{kind=link}
Abstract
1. Introduction
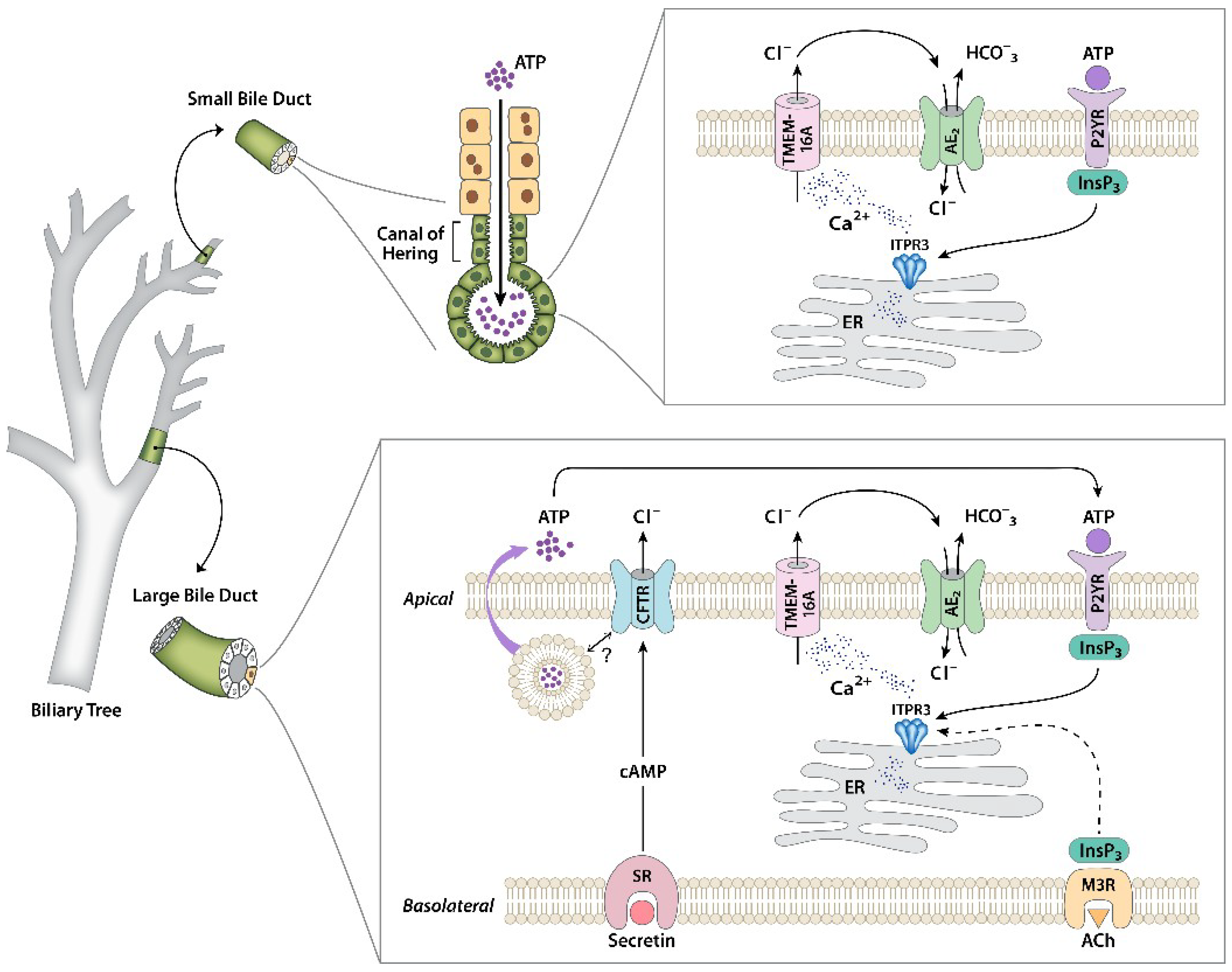
2. Biliary Tree Overview
3. Experimental Models to Study Signaling in Cholangiocytes
4. Regulation of Cholangiocyte Secretory Activity by Calcium Signaling
5. Alterations in Ca2+ Signaling in Cholangiopathies
6. Apoptosis in Cholangiocytes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.A.; Gomes, D.A.; Leite, M.F.; Grant, W.; Zhang, L.; Lam, W.; Cheng, Y.C.; Bennett, A.M.; Nathanson, M.H. Nucleoplasmic calcium is required for cell proliferation. J. Biol. Chem. 2007, 282, 17061–17068. [Google Scholar] [CrossRef]
- Minagawa, N.; Kruglov, E.A.; Dranoff, J.A.; Robert, M.E.; Gores, G.J.; Nathanson, M.H. The anti-apoptotic protein Mcl-1 inhibits mitochondrial Ca2+ signals. J. Biol. Chem. 2005, 280, 33637–33644. [Google Scholar] [CrossRef] [PubMed]
- Gomes, D.A.; Thompson, M.; Souto, N.C.; Goes, T.S.; Goes, A.M.; Rodrigues, M.A.; Gomez, M.V.; Nathanson, M.H.; Leite, M.F. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 2005, 280, 40892–40900. [Google Scholar] [CrossRef]
- Trampert, D.C.; Nathanson, M.H. Regulation of bile secretion by calcium signaling in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1761–1770. [Google Scholar] [CrossRef]
- Hirata, K.; Nathanson, M.H. Bile duct epithelia regulate biliary bicarbonate excretion in normal rat liver. Gastroenterology 2001, 121, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.J.; Nathanson, M.H. Calcium signaling and the secretory activity of bile duct epithelia. Cell Calcium 2014, 55, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.J.; Nathanson, M.H. Calcium signaling in the liver. Compr. Physiol. 2013, 3, 515–539. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004, 127, 1565–1577. [Google Scholar] [CrossRef]
- Spee, B.; Carpino, G.; Schotanus, B.A.; Katoonizadeh, A.; Vander Borght, S.; Gaudio, E.; Roskams, T. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of Wnt and Notch signalling. Gut 2010, 59, 247–257. [Google Scholar] [CrossRef]
- Itoh, T.; Miyajima, A. Liver regeneration by stem/progenitor cells. Hepatology 2014, 59, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Glaser, S.; Robertson, W.; Phinizy, J.; Rodgers, R.; Caligiuri, A.; LeSage, G. Bile acids stimulate proliferative and secretory events in large but not small cholangiocytes. Am. J. Physiol. Liver Physiol. 1997, 273, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Roberts, S.; Kuntz, S.M.; Ueno, Y.; Gubba, S.; Podila, P.V.; LeSage, G.; LaRusso, N.F. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology 1996, 110, 1636–1643. [Google Scholar] [CrossRef]
- Cheung, A.C.; Lorenzo Pisarello, M.J.; LaRusso, N.F. Pathobiology of biliary epithelia. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.M.; Glaser, S.S.; Francis, H.; Phinizy, J.L.; LeSage, G.; Alpini, G. Functional heterogeneity of cholangiocytes. Semin. Liver Dis. 2002, 22, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; LaRusso, N.F. Cholangiocyte primary cilia in liver health and disease. Dev. Dyn. 2008, 237, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; Decaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Decaen, P.G.; Delling, M.; Vien, T.N.; Clapham, D.E. Direct recording and molecular identification of the calcium channel of primary cilia. Nature 2013, 504, 315–318. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Masyuk, A.I.; Splinter, P.L.; Banales, J.M.; Huang, B.Q.; Tietz, P.S.; Masyuk, T.V.; LaRusso, N.F. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc. Natl. Acad. Sci. 2007, 104, 19138–19143. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Masyuk, T.V.; Huang, B.Q.; Banales, J.M.; Lehmann, G.L.; Radtke, B.N.; Stroope, A.; Masyuk, A.I.; Splinter, P.L.; LaRusso, N.F. Activation of Trpv4 Reduces the Hyperproliferative Phenotype of Cystic Cholangiocytes From an Animal Model of ARPKD. Gastroenterology 2010, 139, 304–314.e2. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Gradilone, S.A.; LaRusso, N.F. Calcium Signaling in Cilia and Ciliary-Mediated Intracellular Calcium Signaling: Are They Independent or Coordinated Molecular Events? Hepatology 2014, 60, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, I.M.; Liedtke, W.; Sanderson, M.J.; Valverde, M.A. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc. Natl. Acad. Sci. 2008, 105, 12611–12616. [Google Scholar] [CrossRef] [PubMed]
- Köttgen, M.; Buchholz, B.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T.; et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Spirli, C.; Mariotti, V.; Villani, A.; Fabris, L.; Fiorotto, R.; Strazzabosco, M. Adenylyl cyclase 5 links changes in calcium homeostasis to cAMP-dependent cyst growth in polycystic liver disease. J. Hepatol. 2017, 66, 571–580. [Google Scholar] [CrossRef]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Fabris, L.; Spirli, C.; Strazzabosco, M. Src kinase inhibition reduces inflammatory and cytoskeletal changes in ΔF508 human cholangiocytes and improves cystic fibrosis transmembrane conductance regulator correctors efficacy. Hepatology 2018, 67, 972–988. [Google Scholar] [CrossRef]
- Nathanson, M.H.; Burgstahler, A.D.; Mennone, A.; Boyer, J.L. Characterization of cytosolic Ca2+ signaling in rat bile duct epithelia. Am. J. Physiol. 1996, 34, G86–G96. [Google Scholar] [CrossRef]
- Hirata, K.; Dufour, J.-F.; Shibao, K.; Knickelbein, R.; O’Neill, A.F.; Bode, H.-P.; Cassio, D.; St-Pierre, M.V.; Larusso, N.F.; Leite, M.F.; et al. Regulation of Ca2+ signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms. Hepatology 2002, 36, 284–296. [Google Scholar] [CrossRef]
- Mennone, A.; Alvaro, D.; Cho, W.; Boyer, J.L. Isolation of small polarized bile duct units. Proc. Natl. Acad. Sci. USA 1995, 92, 6527–6531. [Google Scholar] [CrossRef]
- Spirlı̀, C.; Nathanson, M.H.; Fiorotto, R.; Duner, E.; Denson, L.A.; Sanz, J.M.; Di Virgilio, F.; Okolicsanyi, L.; Casagrande, F.; Strazzabosco, M. Proinflammatory Cytokines Inhibit Secretion in Rat Bile Duct Epithelium. Gastroenterology 2001, 121, 156–169. [Google Scholar] [CrossRef]
- Minagawa, N.; Nagata, J.; Shibao, K.; Masyuk, A.I.; Gomes, D.A.; Rodrigues, M.A.; Lesage, G.; Akiba, Y.; Kaunitz, J.D.; Ehrlich, B.E.; et al. Cyclic AMP Regulates Bicarbonate Secretion in Cholangiocytes Through Release of ATP Into Bile. Gastroenterology 2007, 133, 1592–1602. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Gong, A.Y.; Kip, S.; Burke, M.J.; LaRusso, N.F. Perfused rat intrahepatic bile ducts secrete and absorb water, solute, and ions. Gastroenterology 2000, 119, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, J.A.; Masyuk, A.I.; Kruglov, E.A.; Larusso, N.F.; Nathanson, M.H. Polarized expression and function of P2Y ATP receptors in rat bile duct epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Shibao, K.; Hirata, K.; Robert, M.E.; Nathanson, M.H. Loss of Inositol 1,4,5-Triphosphate Receptors From Bile Duct pithelia Is a Common Event in Cholestasis. Gastroenterology 5085, 5085, 1175–1187. [Google Scholar] [CrossRef]
- Joplin, R.; Strain, A.J.; Neuberger, J.M. Immuno-isolation and culture of biliary epithelial cells from normal human liver. In Vitro Cell. Dev. Biol. 1989, 25, 1189–1192. [Google Scholar] [CrossRef]
- Joplin, R.; Strain, A.J.; Neuberger, J.M. Biliary Epithelial Cells From the Liver of Patients With Primary Biliary Cirrhosis : Isolation, Characterization, and Short-Term Culture. J. Pathol. 1990, 162, 255–260. [Google Scholar] [CrossRef]
- Ishii, M.; Vroman, B.; LaRusso, N.F. Isolation and Morphologic Cells From Normal Rat Liver. Gastroenterology 1989, 97, 1236–1247. [Google Scholar] [CrossRef]
- Vroman, B.; LaRusso, N.F. Development and characterization of polarized primary cultures of rat intrahepatic bile duct epithelial cells. Lab Invest. 1996, 74, 303–313. [Google Scholar]
- Banales, J.M.; Sáez, E.; Uriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; LaRusso, N.F. Up-regulation of microRNA 506 leads to decreased Cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef]
- Spirli, C.; Okolicsanyi, S.; Fiorotto, R.; Fabris, L.; Cadamuro, M.; Lecchi, S.; Tian, X.; Somlo, S.; Strazzabosco, M. ERK1/2-Dependent Vascular Endothelial Growth Factor Signaling Sustains Cyst Growth in Polycystin-2 Defective Mice. Gastroenterology 2010, 138, 360–371. [Google Scholar] [CrossRef]
- Fiorotto, R.; Villani, A.; Kourtidis, A.; Scirpo, R.; Amenduni, M.; Geibel, P.J.; Cadamuro, M.; Spirli, C.; Anastasiadis, P.Z.; Strazzabosco, M. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity. Hepatology 2016, 64, 2118–2134. [Google Scholar] [CrossRef]
- Huang, B.Q.; Masyuk, T.V.; Muff, M.A.; Tietz, P.S.; Masyuk, A.I.; LaRusso, N.F. Isolation and characterization of cholangiocyte primary cilia. AJP Gastrointest. Liver Physiol. 2006, 291, G500–G509. [Google Scholar] [CrossRef] [PubMed]
- Dianat, N.; Dubois-Pot-Schneider, H.; Steichen, C.; Desterke, C.; Leclerc, P.; Raveux, A.; Combettes, L.; Weber, A.; Corlu, A.; Dubart-Kupperschmitt, A. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology 2014, 60, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Dianat, N.; Steichen, C.; Vallier, L.; Weber, A.; Dubart-Kupperschmitt, A. Human Pluripotent Stem Cells for Modelling Human Liver Diseases and Cell Therapy. Curr. Gene Ther. 2013, 13, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Ogawa, S.; Bear, C.E.; Ahmadi, S.; Chin, S.; Li, B.; Grompe, M.; Keller, G.; Kamath, B.M.; Ghanekar, A. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; De Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Ghanekar, A.; Kamath, B.M. Cholangiocytes derived from induced pluripotent stem cells for disease modeling. Curr. Opin. Gastroenterol. 2016, 32, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, R.; Spirlì, C.; Fabris, L.; Cadamuro, M.; Okolicsanyi, L.; Strazzabosco, M. Ursodeoxycholic Acid Stimulates Cholangiocyte Fluid Secretion in Mice via CFTR-Dependent ATP Secretion. Gastroenterology 2007, 133, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, M.H.; Burgstahler, A.D.; Masyuk, A.; Larusso, N.F. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem. J. 2001, 358, 1–5. [Google Scholar] [CrossRef]
- Schlosser, S.F.; Burgstahler, A.D.; Nathanson, M.H. Isolated rat hepatocytes can signal to other hepatocytes and bile duct cells by release of nucleotides. Proc. Natl. Acad. Sci. USA 1996, 93, 9948–9953. [Google Scholar] [CrossRef]
- Li, Q.; Dutta, A.; Kresge, C.; Bugde, A.; Feranchak, A.P. Bile acids stimulate cholangiocyte fluid secretion by activation of transmembrane member 16A Cl−channels. Hepatology 2018, 68, 187–199. [Google Scholar] [CrossRef]
- Pusl, T.; Nathanson, M.H. The role of inositol 1,4,5-trisphosphate receptors in the regulation of bile secretion in health and disease. Biochem. Biophys. Res. Commun. 2004, 322, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Franca, A.; Filho, A.C.M.L.; Guerra, M.T.; Weerachayaphorn, J.; Santos, M.L. dos; Njei, B.; Robert, M.; Lima, C.X.; Vidigal, P.V.T.; Banales, J.M.; Ananthanarayanam, M.; et al. Effects of endotoxin on type 3 inositol 1,4,5-trisphosphate receptor in human cholangiocytes. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, M.H.; Fallon, M.B.; Padfield, P.J.; Maranto, A.R. Localization of the type 3 inositol 1,4,5-Trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. J. Biol. Chem. 1994, 269, 4693–4696. [Google Scholar] [PubMed]
- Nagata, J.; Guerra, M.T.; Shugrue, C.A.; Gomes, D.A.; Nagata, N.; Nathanson, M.H. Lipid Rafts Establish Calcium Waves in Hepatocytes. Gastroenterology 2007, 133, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.J.; Oliveira, A.G.; Schroeder, L.K.; Allgeyer, E.S.; Bewersdorf, J.; Nathanson, M.H. Apical localization of inositol 1,4,5-trisphosphate receptors is independent of extended synaptotagmins in hepatocytes. PLoS ONE 2014, 9, e114043. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Nathanson, M.H.; Burgstahler, A.D.; Okazaki, K.; Mattei, E.; Sears, M.L. Relationship between Inositol 1,4,5-Trisphosphate Receptor Isofors and Subcellular Ca2+ Signaling Patterns in Nonpigmented Ciliary Epithelia. Invest. Ophthalmol. Vis. Sci. 1999, 40, 2046–2053. [Google Scholar] [PubMed]
- Hagar, R.E.; Burgstahler, A.D.; Nathanson, M.H.; Ehrlich, B.E. Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature 1998, 396, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Chacón, R.; Königstorfer, A.; Gerber, S.H.; García, J.; Matos, M.F.; Stevens, C.F.; Brose, N.; Rizo, J.; Rosenmund, C.; Südhof, T.C. Synaptotagmin I functions as a calcium regulator of release probability. Nature 2001, 410, 41–49. [Google Scholar] [CrossRef]
- Hu, K.; Carroll, J.; Fedorovich, S.; Rickman, C.; Sukhodub, A.; Davietov, B. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature 2002, 415, 646–650. [Google Scholar] [CrossRef]
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75, 409–418. [Google Scholar] [CrossRef]
- Guerra, M.T.; Nathanson, M.H. Calcium signaling and secretion in cholangiocytes. Pancreatology 2015, 15, S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, N.; Ehrlich, B.E.; Nathanson, M.H. Calcium signaling in cholangiocytes. World J. Gastroenterol. 2006, 12, 3466–3470. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L. Bile duct epithelium: Frontiers in transport physiology. Am. J. Physiol. 1996, 270, G1–G5. [Google Scholar] [CrossRef]
- Alpini, G.; Phinizy, J.L.; Glaser, S.; Francis, H.; Benedetti, A.; Marucci, L.; LeSage, G. Development and characterization of secretin-stimulated secretion of cultured rat cholangiocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G1066–G1073. [Google Scholar] [CrossRef] [PubMed]
- Kanno, N.; LeSage, G.; Glaser, S.; Alpini, G. Regulation of cholangiocyte bicarbonate secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G612–G625. [Google Scholar] [CrossRef] [PubMed]
- Fouassier, L.; Duan, C.; Feranchak, A.P.; Yun, C.H.C.; Sutherland, E.; Simon, F.; Fitz, J.G.; Doctor, R.B. Ezrin-radixin-moesin-binding phosphoprotein 50 is expressed at the apical membrane of rat liver epithelia. Hepatology 2001, 33, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Khimji, A.K.; Kresge, C.; Bugde, A.; Dougherty, M.; Esser, V.; Ueno, Y.; Glaser, S.S.; Alpini, G.; Rockey, D.C.; et al. Identification and functional characterization of TMEM16A, a Ca2+-activated Cl-channel activated by extracellular nucleotides, in biliary epithelium. J. Biol. Chem. 2011, 286, 766–776. [Google Scholar] [CrossRef]
- Weerachayaphorn, J.; Amaya, M.; Spirli, C.; Chansela, P.; Mitchell, K.; Ananthanarayanan, M.; Nathanson, M. Nuclear Factor Erythroid 2-like 2 Regulates Expression of Inositol 1,4,5-trisphosphate Receptor, Type 3 and Calcium Signaling in Cholangiocytes. Gastroenterology 2015, 149, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, M.; Banales, J.M.; Guerra, M.T.; Spirli, C.; Munoz-Garrido, P.; Mitchell-Richards, K.; Tafur, D.; Saez, E.; Nathanson, M.H. Post-translational regulation of the type III inositol 1,4,5-trisphosphate receptor by miRNA-506. J. Biol. Chem. 2015, 290, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Paumgartner, G.; Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology 2002, 36, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Poupon, R.E.; Poupon, R.; Balkau, B. Ursodiol for the long-term treatment of primary biliary cirrhosis. N. Engl. J. Med. 1994, 330, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Francis, H.; Benedetti, A.; Ueno, Y.; Fava, G.; Venter, J.; Reichenbach, R.; Mancino, M.G.; Summers, R.; Alpini, G.; et al. Ca2+-dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. Am. J. Pathol. 2006, 168, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Nakanuma, Y. Detection of apoptosis and expression of apoptosis-related proteins during human intrahepatic bile duct development. Am. J. Pathol. 1995, 146, 67–74. [Google Scholar] [PubMed]
- Bhathal, P.S.; Gall, J.A. Deletion of hyperplastic biliary epithelial cells by apoptosis following removal of the proliferative stimulus. Liver 1985, 5, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.E.; Rust, C.; Eichhorn, P.; Beuers, U.; Holdenrieder, S. Immune-mediated liver diseases: Programmed cell death ligands and circulating apoptotic markers. Expert Rev. Mol. Diagn. 2009, 9, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Erickson, N.; Mohanty, S.K.; Shivakumar, P.; Sabla, G.; Chakraborty, R.; Bezerra, J.A. Temporal-spatial activation of apoptosis and epithelial injury in murine experimental biliary atresia. Hepatology 2008, 47, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kojima, Y.; Ikejima, K.; Harada, K.; Yamashina, S.; Okumura, K.; Aoyama, T.; Frese, S.; Ikeda, H.; Haynes, N.M.; et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc. Natl. Acad. Sci. USA 2008, 105, 10895–10900. [Google Scholar] [CrossRef] [PubMed]
- Szalai, G.; Krishnamurhy, R.; Hajnóczky, G. Apoptosis driven by IP3 -linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Thomas, A.P.; Hajnóczky, G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999, 18, 96–108. [Google Scholar] [CrossRef]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3 -sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Feriod, C.N.; Gustavo Oliveira, A.; Guerra, M.T.; Nguyen, L.; Mitchell Richards, K.; Jurczak, M.J.; Ruan, H.-B.; Paulo Camporez, J.; Yang, X.; Shulman, G.I.; et al. Hepatic inositol 1,4,5 trisphosphate receptor type 1 mediates fatty liver. Hepatol. Commun. 2017, 1, 23–35. [Google Scholar] [CrossRef]
- Li, C.; Fox, C.J.; Master, S.R.; Bindokas, V.P.; Chodosh, L.A.; Thompson, C.B. Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 9830–9835. [Google Scholar] [CrossRef] [PubMed]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Taniai, M.; Grambihler, A.; Higuchi, H.; Werneburg, N.; Bronk, S.F.; Farrugia, D.J.; Kaufmann, S.H.; Gores, G.J. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004, 64, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+ -mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, M.A.; Gomes, D.A.; Nathanson, M.H. Calcium Signaling in Cholangiocytes: Methods, Mechanisms, and Effects. Int. J. Mol. Sci. 2018, 19, 3913. https://doi.org/10.3390/ijms19123913
Rodrigues MA, Gomes DA, Nathanson MH. Calcium Signaling in Cholangiocytes: Methods, Mechanisms, and Effects. International Journal of Molecular Sciences. 2018; 19(12):3913. https://doi.org/10.3390/ijms19123913
Chicago/Turabian StyleRodrigues, Michele Angela, Dawidson Assis Gomes, and Michael Harris Nathanson. 2018. "Calcium Signaling in Cholangiocytes: Methods, Mechanisms, and Effects" International Journal of Molecular Sciences 19, no. 12: 3913. https://doi.org/10.3390/ijms19123913
APA StyleRodrigues, M. A., Gomes, D. A., & Nathanson, M. H. (2018). Calcium Signaling in Cholangiocytes: Methods, Mechanisms, and Effects. International Journal of Molecular Sciences, 19(12), 3913. https://doi.org/10.3390/ijms19123913