Mitochondrial Dynamics in Stem Cells and Differentiation


Abstract
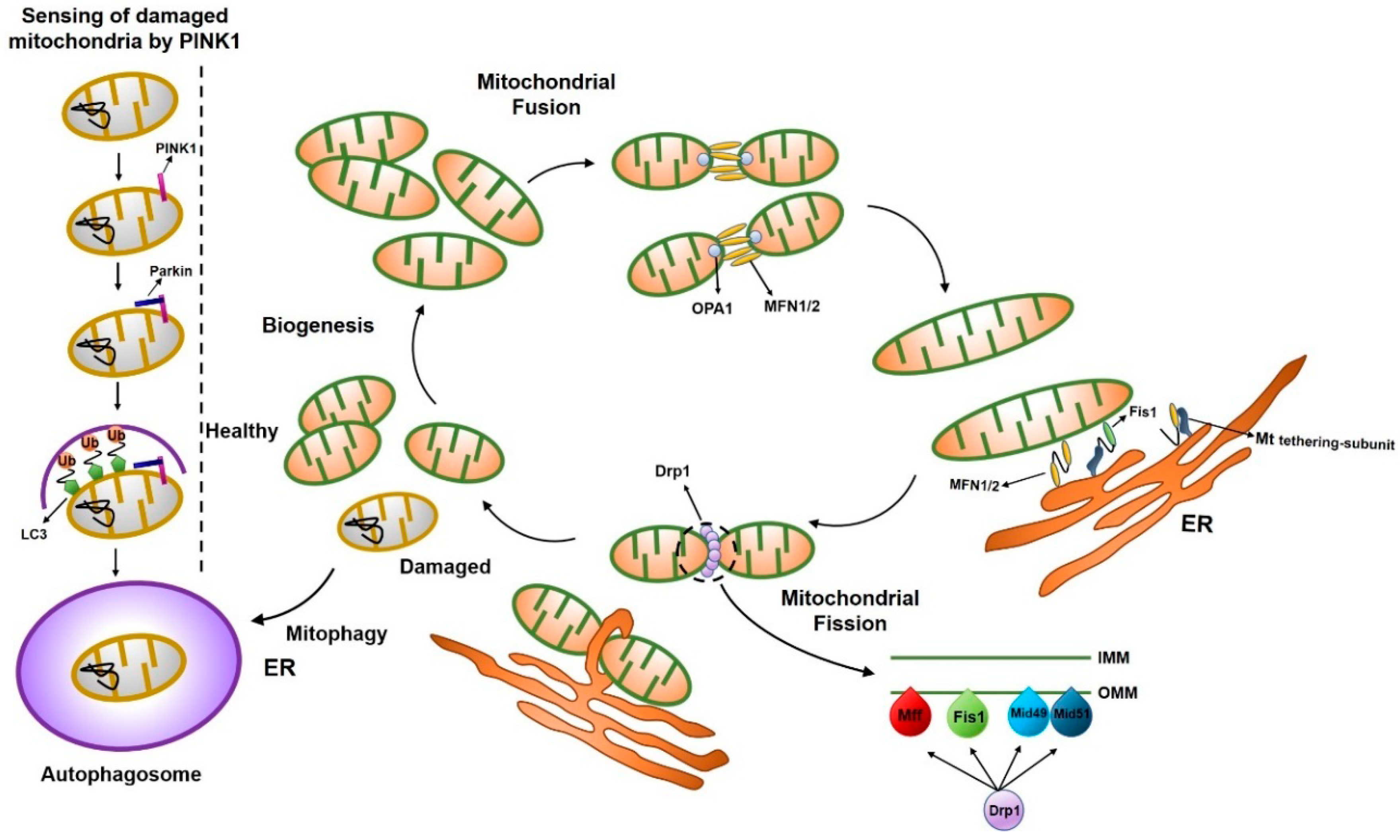
1. Introduction
2. Components Determining the Mitochondrial Structure
2.1. Post-Translational Modification of Mitochondrial Proteins
2.2. Mitochondrial Fission Proteins
2.3. Mitochondrial Fission Accessory Proteins
2.4. Mitochondrial Fusion Proteins
2.5. Mitochondrial Fusion Accessory Proteins
3. Cellular Metabolism and Mitochondrial Dynamics
3.1. The Mitochondrial Morphologies and Energy Metabolism in Various Stem Cells
3.2. Warburg Effect: Survival Strategy of Proliferative Glycolytic Cells
3.3. Metabolic Regulation in Mitochondria for the Maintenance of the Pluripotent State
4. Mitochondrial Dynamics in the Reprogramming Process
4.1. Mitochondrial Fission Proteins Affect Pluripotential Reprogramming
4.2. Mitochondrial Fusion Proteins Affect Pluripotential Reprogramming
5. Mitochondrial Dynamics in the Differentiation Process
5.1. Mitochondrial Fission Proteins Affect Cellular Differentiation
5.2. Mitochondrial Fusion Proteins Affect Cellular Differentiation
6. Mitochondrial Dynamics in Aging
6.1. Mitochondrial ROS Impact on Cellular Senescence-Related Mitochondrial Dynamics
6.2. Mitochondrial Involvement in Cell Death Activation and Calcium Signaling
7. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OMM | Outer mitochondrial membrane |
| IMM | Inner mitochondrial membrane |
| OXPHOS | Oxidative phosphorylation |
| ESC | Embryonic stem cell |
| EpiSC | Epiblast stem cell |
| NSC | Neural stem cell |
| NPC | Neural progenitor cell |
| MSC | Mesenchymal stem cell |
| HSC | Hematopoietic stem cell |
| HPC | Hematopoietic progenitor cell |
| PSC | Pluripotent stem cell |
| iPSC | Induced pluripotent stem cell |
| MEF | Mouse embryonic fibroblast |
| HUVEC | Human endothelial cell |
References
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, K.S.; Scorrano, L. (De)constructing mitochondria: What for? Physiology 2006, 21, 233–241. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. An intimate liaison: Spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.L.; Sasaki, Y.; Tuck, E.; Milbrandt, J.; Baloh, R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012, 32, 4145–4155. [Google Scholar] [CrossRef] [PubMed]
- Dunwoodie, S.L. The role of hypoxia in development of the Mammalian embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Morrow, R.M.; Picard, M.; Derbeneva, O.; Leipzig, J.; McManus, M.J.; Gouspillou, G.; Barbat-Artigas, S.; Dos Santos, C.; Hepple, R.T.; Murdock, D.G.; et al. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C. Mitochondrial metabolites: Undercover signalling molecules. Interface Focus 2017, 7, 20160100. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics-fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Lewis, M.R.; Lewis, W.H. Mitochondria (and other cytoplasmic structures) in tissue cultures. Am. J. Anat. 1915, 17, 339–401. [Google Scholar] [CrossRef]
- Okamoto, K.; Shaw, J.M. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005, 39, 503–536. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [PubMed]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Harder, Z.; Zunino, R.; McBride, H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 2004, 14, 340–345. [Google Scholar] [CrossRef]
- Wasiak, S.; Zunino, R.; McBride, H.M. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 2007, 177, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; Zunino, R.; McBride, H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef]
- Zunino, R.; Schauss, A.; Rippstein, P.; Andrade-Navarro, M.; McBride, H.M. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J. Cell Sci. 2007, 120, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef]
- Hanover, J.A.; Yu, S.; Lubas, W.B.; Shin, S.H.; Ragano-Caracciola, M.; Kochran, J.; Love, D.C. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch. Biochem. Biophys. 2003, 409, 287–297. [Google Scholar] [CrossRef]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Hu, Y.; Oyeleye, M.O.; Dillmann, W.H. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J. Biol. Chem. 2009, 284, 547–555. [Google Scholar] [CrossRef]
- Pinho, T.S.; Correia, S.C.; Perry, G.; Ambrosio, A.F.; Moreira, P.I. Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. Biochim. Biophys. Acta Mol. Basis Dis. 2018. [Google Scholar] [CrossRef]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Mozdy, A.D.; McCaffery, J.M.; Shaw, J.M. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef]
- Otsuga, D.; Keegan, B.R.; Brisch, E.; Thatcher, J.W.; Hermann, G.J.; Bleazard, W.; Shaw, J.M. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J. Cell Biol. 1998, 143, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Naylor, K.; Ingerman, E.; Okreglak, V.; Marino, M.; Hinshaw, J.E.; Nunnari, J. Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J. Biol. Chem. 2006, 281, 2177–2183. [Google Scholar] [CrossRef] [PubMed]
- Karren, M.A.; Coonrod, E.M.; Anderson, T.K.; Shaw, J.M. The role of Fis1p-Mdv1p interactions in mitochondrial fission complex assembly. J. Cell Biol. 2005, 171, 291–301. [Google Scholar] [CrossRef]
- Tieu, Q.; Nunnari, J. MDV1P is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 2000, 151, 353–366. [Google Scholar] [CrossRef]
- Lackner, L.L.; Nunnari, J.M. The molecular mechanism and cellular functions of mitochondrial division. Biochim. Biophys. Acta 2009, 1792, 1138–1144. [Google Scholar] [CrossRef]
- Lackner, L.L.; Horner, J.S.; Nunnari, J. Mechanistic analysis of a dynamin effector. Science 2009, 325, 874–877. [Google Scholar] [CrossRef]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2011, 2, 167–172. [Google Scholar] [CrossRef]
- Yu, R.; Liu, T.; Jin, S.B.; Ning, C.; Lendahl, U.; Nister, M.; Zhao, J. MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci. Rep. 2017, 7, 880. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlen, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nister, M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef]
- Wagner, K.M.; Ruegg, M.; Niemann, A.; Suter, U. Targeting and function of the mitochondrial fission factor GDAP1 are dependent on its tail-anchor. PLoS ONE 2009, 4, e5160. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Valdes-Sanchez, T.; Sanchez-Piris, M.; Sirkowski, E.E.; Scherer, S.S.; Farinas, I.; Palau, F. Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease. J. Cell. Mol. Med. 2008, 12, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, J.; Chevrollier, A.; Bonneau, D.; Verny, C.; Procaccio, V.; Reynier, P.; Ferre, M. A locus-specific database for mutations in GDAP1 allows analysis of genotype-phenotype correlations in Charcot-Marie-Tooth diseases type 4A and 2K. Orphanet J. Rare Dis. 2011, 6, 87. [Google Scholar] [CrossRef]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S.; Igarashi, Y. Functional role of glycosphingolipids in cell recognition and signaling. J. Biochem. 1995, 118, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Modregger, J.; Schmidt, A.A.; Ritter, B.; Huttner, W.B.; Plomann, M. Characterization of Endophilin B1b, a brain-specific membrane-associated lysophosphatidic acid acyl transferase with properties distinct from endophilin A1. J. Biol. Chem. 2003, 278, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, J.E. Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 2000, 16, 483–519. [Google Scholar] [CrossRef]
- Schmidt, A.; Wolde, M.; Thiele, C.; Fest, W.; Kratzin, H.; Podtelejnikov, A.V.; Witke, W.; Huttner, W.B.; Soling, H.D. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature 1999, 401, 133–141. [Google Scholar] [CrossRef]
- Gad, H.; Ringstad, N.; Low, P.; Kjaerulff, O.; Gustafsson, J.; Wenk, M.; Di Paolo, G.; Nemoto, Y.; Crun, J.; Ellisman, M.H.; et al. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron 2000, 27, 301–312. [Google Scholar] [CrossRef]
- Huttner, W.B.; Schmidt, A. Lipids, lipid modification and lipid-protein interaction in membrane budding and fission—Insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr. Opin. Neurobiol. 2000, 10, 543–551. [Google Scholar] [CrossRef]
- Cuddeback, S.M.; Yamaguchi, H.; Komatsu, K.; Miyashita, T.; Yamada, M.; Wu, C.; Singh, S.; Wang, H.G. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J. Biol. Chem. 2001, 276, 20559–20565. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Karbowski, M.; Yamaguchi, H.; Kazi, A.; Wu, J.; Sebti, S.M.; Youle, R.J.; Wang, H.G. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 2005, 25, 9369–9382. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Jeong, S.Y.; Youle, R.J. Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol. 2004, 166, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L. Keeping mitochondria in shape: A matter of life and death. Eur. J. Clin. Investig. 2013, 43, 886–893. [Google Scholar] [CrossRef]
- Malka, F.; Guillery, O.; Cifuentes-Diaz, C.; Guillou, E.; Belenguer, P.; Lombes, A.; Rojo, M. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005, 6, 853–859. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef]
- Mozdy, A.D.; Shaw, J.M. A fuzzy mitochondrial fusion apparatus comes into focus. Nat. Rev. Mol. Cell Biol. 2003, 4, 468–478. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2: A mitochondria-shaping protein with signaling roles beyond fusion. Antioxid. Redox Signal. 2008, 10, 621–633. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef] [PubMed]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Rabl, R.; Soubannier, V.; Scholz, R.; Vogel, F.; Mendl, N.; Vasiljev-Neumeyer, A.; Korner, C.; Jagasia, R.; Keil, T.; Baumeister, W.; et al. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J. Cell Biol. 2009, 185, 1047–1063. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Kitsis, R.N. The mitochondrial dynamism-mitophagy-cell death interactome: Multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2010, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Andersen, J.K. Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 2008, 1780, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Dawson, V.L.; Dawson, T.M. Activation mechanisms of the E3 ubiquitin ligase parkin. Biochem. J. 2017, 474, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lammermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Zick, M.; Rabl, R.; Reichert, A.S. Cristae formation-linking ultrastructure and function of mitochondria. Biochim. Biophys. Acta 2009, 1793, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R.; Ebert, B.; Schaffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 1518. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.; Kowaltowski, A.J. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells 2016, 34, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, H.W. Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell biol. 2017, 49, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, L.L.; de Almeida, M.J.; Corrigan, D.J.; Mumau, M.; Snoeck, H.W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 2016, 529, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef]
- Zhang, H.; Badur, M.G.; Divakaruni, A.S.; Parker, S.J.; Jager, C.; Hiller, K.; Murphy, A.N.; Metallo, C.M. Distinct Metabolic States Can Support Self-Renewal and Lipogenesis in Human Pluripotent Stem Cells under Different Culture Conditions. Cell Rep. 2016, 16, 1536–1547. [Google Scholar] [CrossRef]
- Choi, H.W.; Kim, J.H.; Chung, M.K.; Hong, Y.J.; Jang, H.S.; Seo, B.J.; Jung, T.H.; Kim, J.S.; Chung, H.M.; Byun, S.J.; et al. Mitochondrial and metabolic remodeling during reprogramming and differentiation of the reprogrammed cells. Stem Cells Dev. 2015, 24, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Suhr, S.T.; Chang, E.A.; Tjong, J.; Alcasid, N.; Perkins, G.A.; Goissis, M.D.; Ellisman, M.H.; Perez, G.I.; Cibelli, J.B. Mitochondrial rejuvenation after induced pluripotency. PLoS ONE 2010, 5, e14095. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Cufi, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Vellon, L.; Menendez, J.A. Mitochondrial fusion by pharmacological manipulation impedes somatic cell reprogramming to pluripotency: New insight into the role of mitophagy in cell stemness. Aging 2012, 4, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Son, M.Y.; Choi, H.; Han, Y.M.; Cho, Y.S. Unveiling the critical role of REX1 in the regulation of human stem cell pluripotency. Stem Cells 2013, 31, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef]
- Prieto, J.; Leon, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; Lopez-Garcia, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Prieto, J.; Leon, M.; Ponsoda, X.; Garcia-Garcia, F.; Bort, R.; Serna, E.; Barneo-Munoz, M.; Palau, F.; Dopazo, J.; Lopez-Garcia, C.; et al. Dysfunctional mitochondrial fission impairs cell reprogramming. Cell Cycle 2016, 15, 3240–3250. [Google Scholar] [CrossRef]
- Son, M.J.; Kwon, Y.; Son, M.Y.; Seol, B.; Choi, H.S.; Ryu, S.W.; Choi, C.; Cho, Y.S. Mitofusins deficiency elicits mitochondrial metabolic reprogramming to pluripotency. Cell Death Differ. 2015, 22, 1957–1969. [Google Scholar] [CrossRef]
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sanchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Sesaki, H.; Kageyama, Y.; Reddy, P.H. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim. Biophys. Acta 2012, 1822, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Shaker, M.R.; Cho, B.; Cho, H.M.; Kim, H.; Kim, J.Y.; Sun, W. Dynamin-related protein 1 controls the migration and neuronal differentiation of subventricular zone-derived neural progenitor cells. Sci. Rep. 2015, 5, 15962. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Falcone, S.; Pisoni, S.; Cipolat, S.; Panzeri, C.; Pambianco, S.; Pisconti, A.; Allevi, R.; Bassi, M.T.; Cossu, G.; et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 2010, 17, 1684–1696. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef]
- Sin, J.; Andres, A.M.; Taylor, D.J.R.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef]
- Hoque, A.; Sivakumaran, P.; Bond, S.T.; Ling, N.X.Y.; Kong, A.M.; Scott, J.W.; Bandara, N.; Hernandez, D.; Liu, G.S.; Wong, R.C.B.; et al. Mitochondrial fission protein Drp1 inhibition promotes cardiac mesodermal differentiation of human pluripotent stem cells. Cell Death Discov. 2018, 4, 39. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100. [Google Scholar] [CrossRef]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.B.; Jeffery, I.B.; O’Toole, P.W. The role of the microbiota in ageing: Current state and perspectives. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Shadyab, A.H.; LaCroix, A.Z. Genetic factors associated with longevity: A review of recent findings. Ageing Res. Rev. 2015, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sergiev, P.V.; Dontsova, O.A.; Berezkin, G.V. Theories of aging: An ever-evolving field. Acta Nat. 2015, 7, 9–18. [Google Scholar]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef]
- Lin, J.R.; Shen, W.L.; Yan, C.; Gao, P.J. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Lang, A.; Anand, R.; Altinoluk-Hambuchen, S.; Ezzahoini, H.; Stefanski, A.; Iram, A.; Bergmann, L.; Urbach, J.; Bohler, P.; Hansel, J.; et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging 2017, 9, 2163–2189. [Google Scholar] [CrossRef] [PubMed]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef]
- Kaddour-Djebbar, I.; Choudhary, V.; Brooks, C.; Ghazaly, T.; Lakshmikanthan, V.; Dong, Z.; Kumar, M.V. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. Int. J. Oncol. 2010, 36, 1437–1444. [Google Scholar] [PubMed]
- Wang, Y.; Subramanian, M.; Yurdagul, A., Jr.; Barbosa-Lorenzi, V.C.; Cai, B.; de Juan-Sanz, J.; Ryan, T.A.; Nomura, M.; Maxfield, F.R.; Tabas, I. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 2017, 171, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Slack, R.S. The phosphorylation state of Drp1 determines cell fate. EMBO Rep. 2007, 8, 912–913. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L. Multiple functions of mitochondria-shaping proteins. Nov. Found. Symp. 2007, 287, 47–55. [Google Scholar]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Parasuraman, R.; Greenwood, P.M.; Alexander, G.E. Alzheimer disease constricts the dynamic range of spatial attention in visual search. Neuropsychologia 2000, 38, 1126–1135. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; Garcia-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marin, I.; Vilchez, J.J.; Palau, F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Le, W.; Shahed, J.; Xie, W.; Jankovic, J. Mutation analysis of the parkin and PINK1 genes in American Caucasian early-onset Parkinson disease families. Neurosci. Lett. 2008, 430, 18–22. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Li, G.; Wang, J.; Yang, E.S. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J. Mol. Neurosci. 2008, 34, 165–171. [Google Scholar] [CrossRef]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat.Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Component | Location | Size | Function | Reference |
|---|---|---|---|---|
| Drp1 | OMM | 80 kDa | Fission main protein | [40,99] |
| Fis1 | OMM | 18 kDa | Fission accessory protein | [43,99] |
| Mff | OMM | 55 kDa | Fission accessory protein | [56,99] |
| MiD51 | OMM | 51 kDa | Fission accessory protein | [59,99] |
| MiD49 | OMM | 49 kDa | Fission accessory protein | [59,99] |
| GDAP1 | OMM | 16~21 kDa | Fission accessory protein | [63] |
| Endophilin | Intracellular membrane | 40~43 kDa | Fission accessory protein | [66] |
| Opa1 | IMM | 86 kDa | Fission/Fusion protein | [21] |
| Mfn1 | IMM | 84 kDa | Fusion main protein | [19] |
| Mfn2 | IMM | 86.1 kDa | Fusion main protein | [19] |
| PINK1 | IMM | 63 kDa | Fusion accessory protein | [90] |
| PARKIN | IMM | 52 kDa | Fusion accessory protein | [90] |
| Cell type | Potency | Morphology | Predominant Energy Metabolism | Reference |
|---|---|---|---|---|
| Embryonic stem cells (ESCs) | Naïve pluripotency | Non-fused spherical | Glycolysis (Higher OXPHOS than EpiSCs) | [104] |
| Epiblast stem cells (EpiSCs) | Primed pluripotency | Non-fused spherical | Glycolysis | [104] |
| Neural stem cells (NSCs) | Multipotency | Fused elongated | Glycolysis | [105,106] |
| Neural progenitor cells (NPCs) | Multipotency | Non-fused spherical | Glycolysis | [105,106] |
| Neurons | - | Fused elongated | OXPHOS | [105,106] |
| Mesenchymal stem cells (MSCs) | Multipotency | Fused elongated | Glycolysis | [107] |
| Hematopoietic stem cells (HSCs) | Multipotency | Fused elongated | Glycolysis | [108] |
| Hematopoietic progenitor cells (HPCs) | Multipotency | Fused elongated | Glycolysis (Higher OXPHOS than HSCs) | [109] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. https://doi.org/10.3390/ijms19123893
Seo BJ, Yoon SH, Do JT. Mitochondrial Dynamics in Stem Cells and Differentiation. International Journal of Molecular Sciences. 2018; 19(12):3893. https://doi.org/10.3390/ijms19123893
Chicago/Turabian StyleSeo, Bong Jong, Sang Hoon Yoon, and Jeong Tae Do. 2018. "Mitochondrial Dynamics in Stem Cells and Differentiation" International Journal of Molecular Sciences 19, no. 12: 3893. https://doi.org/10.3390/ijms19123893
APA StyleSeo, B. J., Yoon, S. H., & Do, J. T. (2018). Mitochondrial Dynamics in Stem Cells and Differentiation. International Journal of Molecular Sciences, 19(12), 3893. https://doi.org/10.3390/ijms19123893