Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins

Abstract
1. Introduction
2. Results and Discussion
2.1. Classification of Eukaryotic Proteins by Subcellular Localizations
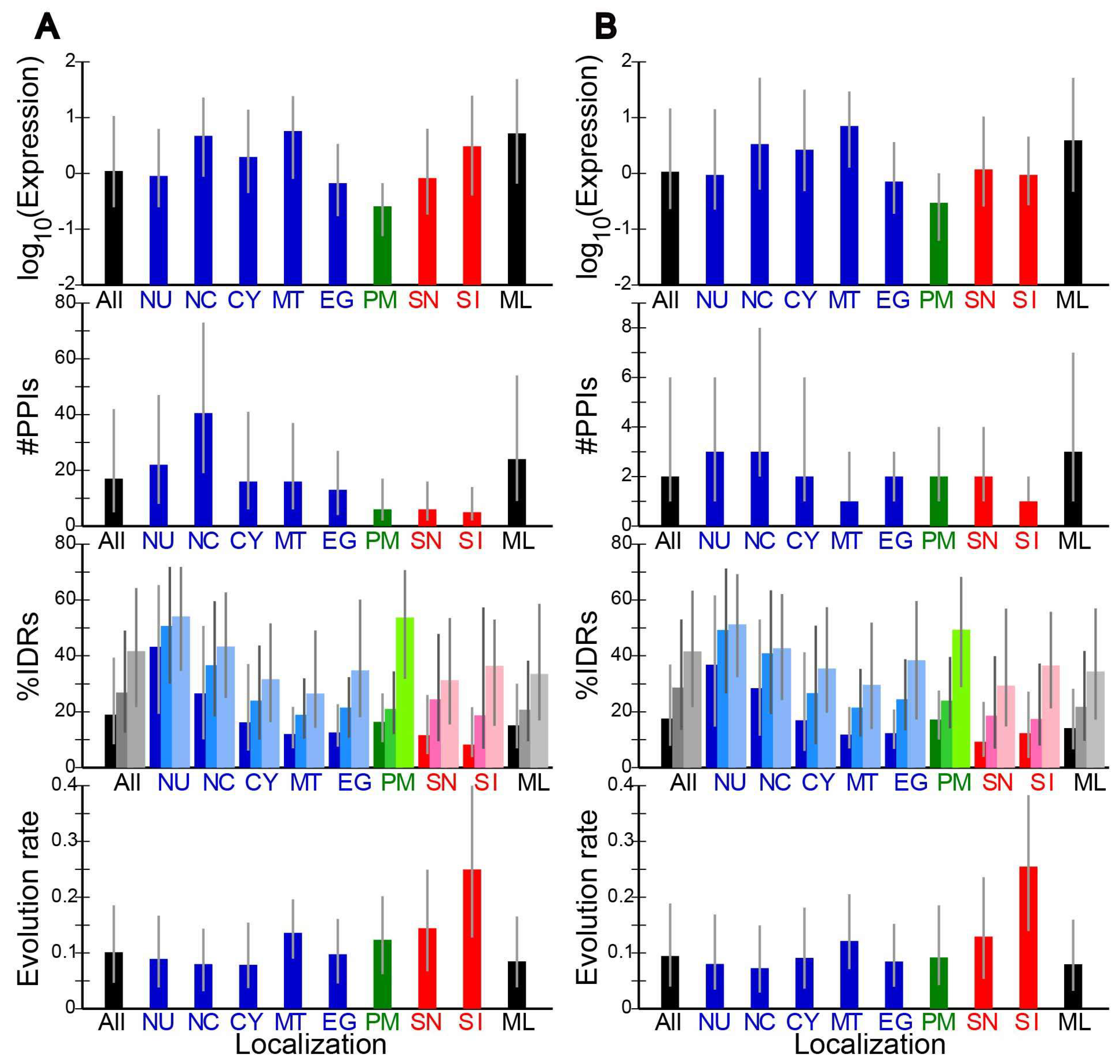
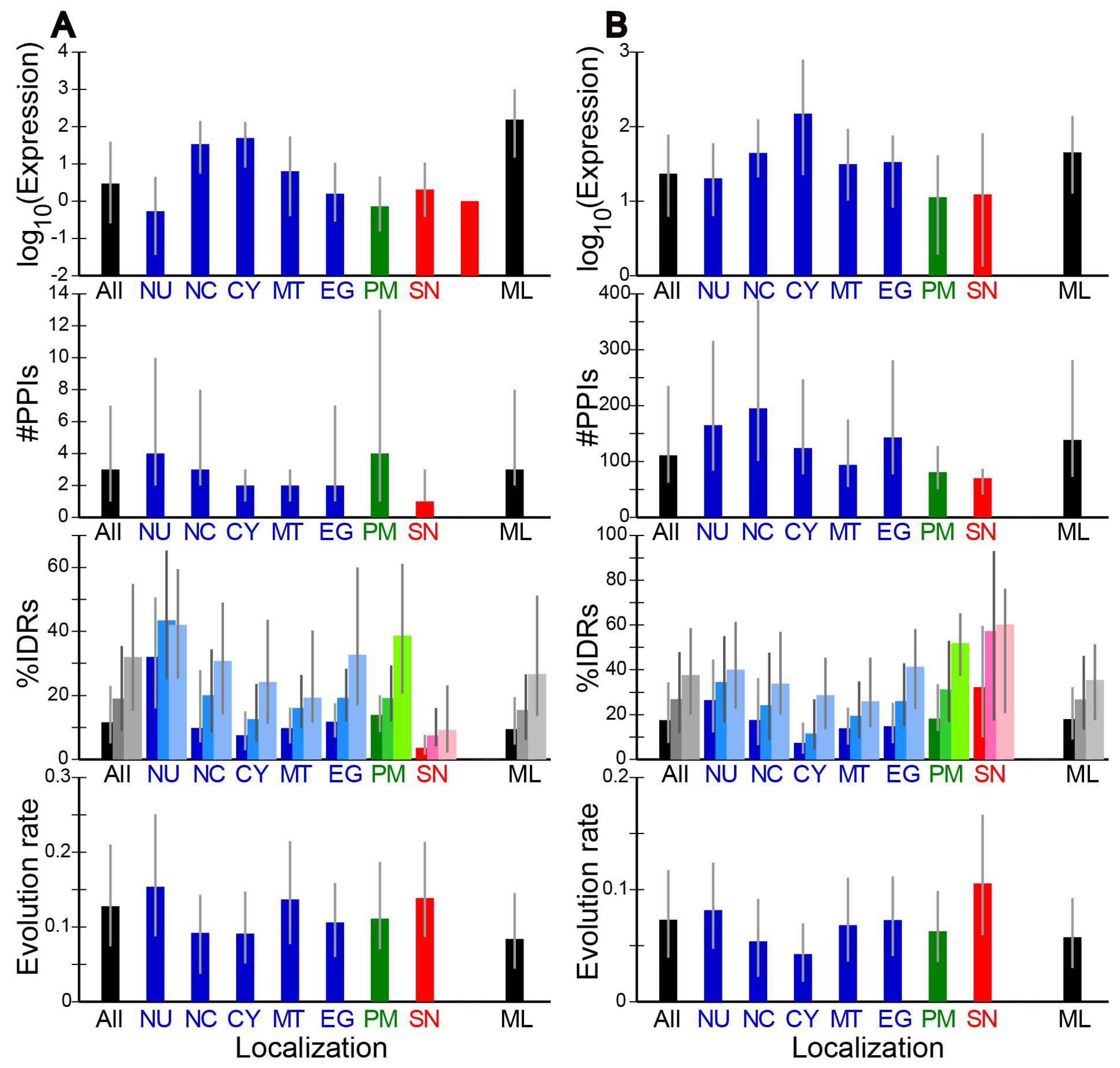
2.2. Evolution Rates and Other Properties of Proteins in Different Subcellular Localizations
2.3. Correlation of Evolution Rates with Protein Properties
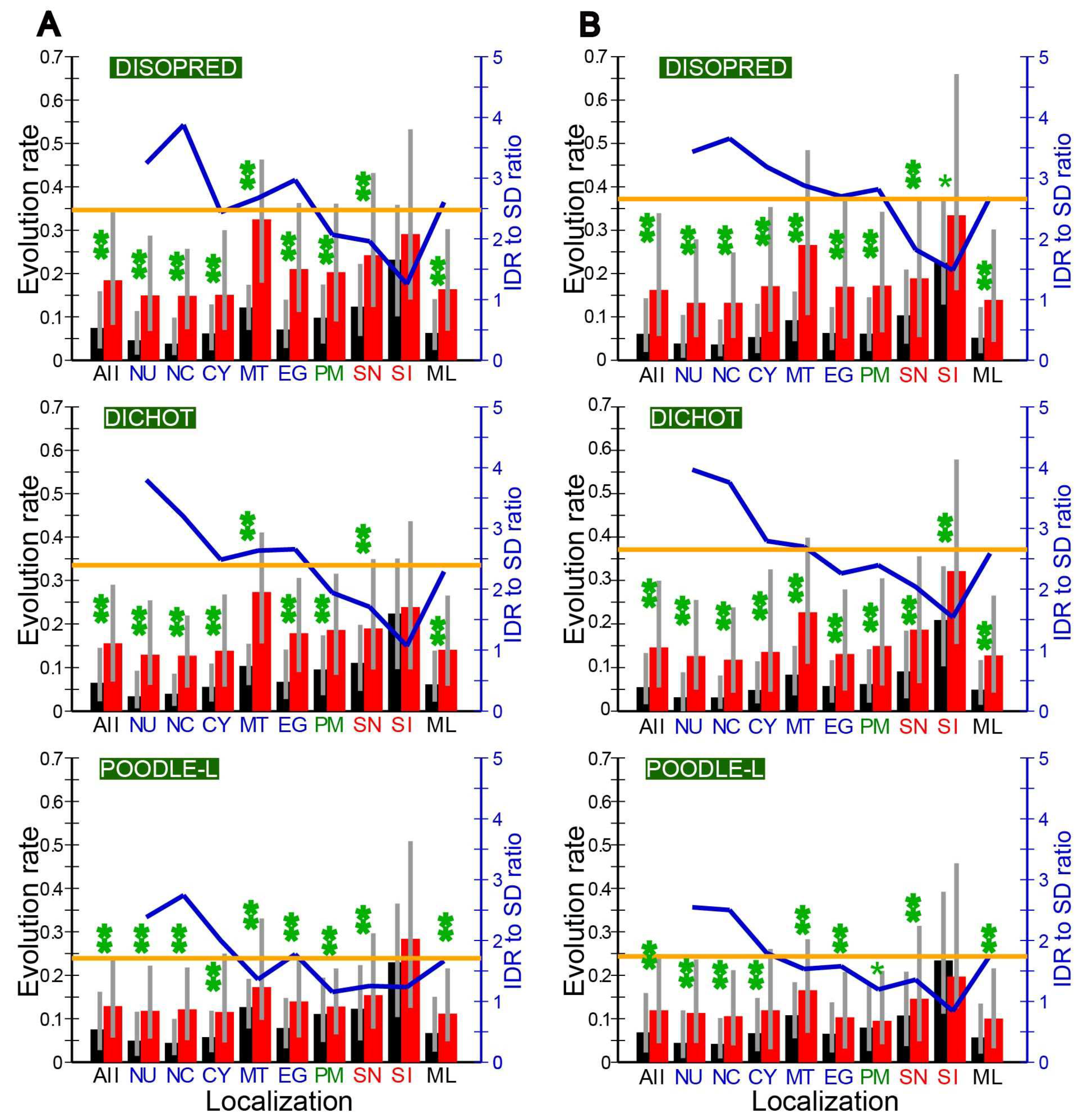
2.4. Evolution Rates in SDs and IDRs in Different Subcellular Localizations
2.5. Examles of Proteins with Nonsynonymous and Synonymous Substitutions
2.6. Significance and Remaining Issues
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
Abbreviations
| SD | Structural domain |
| IDR | Intrinsically disordered region |
| PPI | Protein-to-protein interaction |
| NU | Nucleus |
| NC | Nucleus and cytosol |
| CY | Cytosol |
| MT | Mitochondria |
| EG | Endoplasmic reticulum or Golgi apparatus |
| PM | Plasma membrane |
| SN | Secreted, non-immune-related |
| SI | Secreted, immune-related |
| ML | Multiple localizations except NC |
| dN | Nonsynonymous substitution rate |
| dS | Synonymous substitution rate |
| GO | Gene Ontology |
References
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.B.; Hirsh, A.E.; Steinmetz, L.M.; Scharfe, C.; Feldman, M.W. Evolutionary rate in the protein interaction network. Science 2002, 296, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Krylov, D.M.; Wolf, Y.I.; Rogozin, I.B.; Koonin, E.V. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 2003, 13, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Pál, C.; Papp, B.; Hurst, L.D. Highly expressed genes in yeast evolve slowly. Genetics 2001, 158, 927–931. [Google Scholar] [PubMed]
- Subramanian, S.; Kumar, S. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 2004, 168, 373–381. [Google Scholar] [CrossRef]
- Liu, J.; Perumal, N.E.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006, 359, 1137–1149. [Google Scholar] [CrossRef]
- Homma, K.; Fukuchi, S.; Nishikawa, K.; Sakamoto, S.; Sugawara, H. Intrinsically disordered regions have specific functions in mitochondrial and nuclear proteins. Mol. Biosyst. 2012, 8, 247–255. [Google Scholar] [CrossRef]
- Ito, M.; Tohsato, Y.; Sugisawa, H.; Kohara, S.; Fukuchi, S.; Nishikawa, I.; Nishikawa, K. Intrinsically disordered proteins in human mitochondria. Genes Cells 2012, 17, 817–825. [Google Scholar] [CrossRef]
- Julenius, K.; Pedersen, A.G. Protein evolution is faster outside the cell. Mol. Biol. Evol. 2006, 23, 2039–2048. [Google Scholar] [CrossRef]
- Liao, B.Y.; Weng, M.P.; Zhang, J. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol. Evol. 2010, 2, 39–43. [Google Scholar] [CrossRef]
- Hughes, A.L.; Nei, M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 1988, 335, 167–170. [Google Scholar] [CrossRef]
- Hughes, A.L.; Nei, M. Nucleotide substitution at major histocompatibility complex class II loci: Evidence for overdominant selection. Proc. Natl. Acad. Sci. USA 1989, 86, 958–962. [Google Scholar] [CrossRef]
- Patil, A.; Hughes, A.L.; Zhang, G. Rapid evolution and diversification of mammalian α-defensins as revealed by comparative analysis of rodent and primate genes. Physiol. Genom. 2004, 20, 1–11. [Google Scholar] [CrossRef]
- Morrison, G.M.; Semple, C.A.M.; Kilanowski, F.M.; Hill, R.E.; Dorin, J.R. Signal sequence conservation and mature peptide divergence within subgroups of the murine β-defensin gene family. Mol. Biol. Evol. 2003, 20, 460–470. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Pontillo, A.; Puzzi, L.; Antcheva, N.; Segat, L.; Pacor, S.; Crovella, S.; Tossi, A. Evolution of the primate cathelicidin. Correlation between structural variations and antimicrobial activity. J. Biol. Chem. 2006, 281, 19861–19871. [Google Scholar] [CrossRef]
- Baxt, L.A.; Garza-Mayers, A.C.; Goldberg, M.B. Bacterial subversion of host innate immune pathways. Science 2013, 340, 697–701. [Google Scholar] [CrossRef]
- Sánchez, B.; Urdaci, M.C.; Margolles, A. Extracellular proteins secreted by probiotic bacteria as mediators of effects that promote mucosa-bacteria interactions. Microbiology 2010, 156, 3232–3242. [Google Scholar] [CrossRef]
- Nobre, T.M.; Martynowicz, M.W.; Andreev, K.; Kuzmenko, I.; Nikaido, H.; Gidalevitz, D. Modification of Salmonella lipopolysaccharides prevents the outer membrane penetration of novobiocin. Biophys. J. 2015, 109, 2537–2545. [Google Scholar] [CrossRef]
- Horlick, R.A.; Macomber, J.L.; Bowers, P.M.; Nebern, T.Y.; Tomlinson, G.L.; Krapf, I.P.; Dalton, J.L.; Verdino, P.; King, D.J. Simultaneous surface display and secretion of proteins from mammalian cells facilitate efficient in vitro selection and maturation of antibodies. J. Biol. Chem. 2013, 288, 19861–19869. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Burgess, A.W. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor (1). N. Engl. J. Med. 1992, 327, 28–35. [Google Scholar] [CrossRef]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589, 1251–1258. [Google Scholar] [CrossRef]
- Lucas, A.; McFadden, G. Secreted immunomodulatory viral proteins as novel biotherapeutics. J. Immunol. 2004, 173, 4765–4774. [Google Scholar] [CrossRef]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef]
- Bonin-Debs, A.L.; Boche, I.; Gille, H.; Brinkmann, U. Development of secreted proteins as biotherapeutic agents. Expert Opin. Biol. Ther. 2004, 4, 551–558. [Google Scholar] [CrossRef]
- Castillo-Davis, C.I.; Kondrashov, F.A.; Hartl, D.L.; Kulathinal, R.J. The functional genomic distribution of protein divergence in two animal phyla: Coevolution, genomic conflict, and constraint. Genome Res. 2004, 14, 802–811. [Google Scholar] [CrossRef]
- Ota, M.; Gonja, H.; Koike, R.; Fukuchi, S. Multiple-localization and hub proteins. PLoS ONE 2016, 11, e0156455. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Fukuchi, S.; Homma, K.; Minezaki, Y.; Gojobori, T.; Nishikawa, K. Development of an accurate classification system of proteins into structured and unstructured regions that uncovers novel structural domains: Its application to human transcription factors. BMC Struct. Biol. 2009, 9, 26. [Google Scholar] [CrossRef]
- Hirose, S.; Shimizu, K.; Kanai, S.; Kuroda, Y.; Noguchi, T. POODLE-L: A two-level SVM prediction system for reliably predicting long disordered regions. Bioinformatics 2007, 23, 2046–2053. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ota, T.; Vacquier, V.D. Positive selection is a general phenomenon in the evolution of abalone sperm lysin. Mol. Biol. Evol. 1995, 12, 231–238. [Google Scholar] [CrossRef]
- Swanson, W.J.; Vacquier, V.D. Extraordinary divergence and positive Darwinian selection in a fusagenic protein coating the acrosomal process of abalone spermatozoa. Proc. Natl. Acad. Sci. USA 1995, 92, 4957–4961. [Google Scholar] [CrossRef]
- Tsauer, S.-C.; Wu, C.-I. Positive selection and the molecular evolution of a gene of male reproduction, Acp26Aa, of Drosophila. Mol. Biol. Evol. 1997, 14, 544–549. [Google Scholar] [CrossRef]
- Wyckoff, G.J.; Wang, W.; Wu, C.-I. Rapid evolution of male reproductive genes in the descent of man. Nature 2000, 403, 304–309. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Skrzypek, M.S.; Nash, R.S.; Wong, E.D.; MacPherson, K.A.; Hellerstedt, S.T.; Engel, S.R.; Karra, K.; Weng, S.; Sheppard, T.K.; Binkley, G.; et al. Saccharomyces genome database informs human biology. Nucleic Acids Res. 2018, 46, D736–D742. [Google Scholar] [CrossRef]
- Gramates, L.S.; Marygold, S.J.; dos Santos, G.; Urbano, J.M.; Antonazzo, G.; Matthews, B.B.; Rey, A.J.; Tabone, C.J.; Crosby, M.A.; Emmert, D.B.; et al. FlyBase at 25: Looking to the future. Nucleic Acids Res. 2017, 45, D663–D671. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef]
- Yang, Z. PAML4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDB: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | All | NU | NC | CY | MT | EG | PM | SN | SI | ML |
|---|---|---|---|---|---|---|---|---|---|---|
| H. sapiens | 10,348 | 1639 | 632 | 455 | 377 | 400 | 1116 | 584 | 139 | 3023 |
| M. musculus | 10,068 | 1719 | 546 | 224 | 426 | 514 | 998 | 796 | 125 | 2787 |
| A. thaliana | 8910 | 1032 | 163 | 331 | 356 | 348 | 534 | 431 | 6 | 594 |
| S. cerevisiae | 5304 | 1532 | 232 | 241 | 639 | 458 | 281 | 69 | 0 | 416 |
| Correlation with | H. sapiens | M. musculus | A. thaliana | S. cerevisiae |
|---|---|---|---|---|
| #PPI with ω | −0.293 | −0.194 | −0.054 | −0.195 |
| Expression level with ω | −0.264 | −0.231 | −0.337 | −0.459 |
| %IDR (DISOPRED) with ω | 0.093 | 0.094 | 0.168 | 0.264 |
| %IDR (DICHOT) with ω | 0.113 | 0.146 | 0.052 | 0.303 |
| %IDR (POODLE) with ω | 0.096 | 0.097 | 0.113 | 0.179 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homma, K.; Anbo, H.; Noguchi, T.; Fukuchi, S. Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins. Int. J. Mol. Sci. 2018, 19, 3860. https://doi.org/10.3390/ijms19123860
Homma K, Anbo H, Noguchi T, Fukuchi S. Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins. International Journal of Molecular Sciences. 2018; 19(12):3860. https://doi.org/10.3390/ijms19123860
Chicago/Turabian StyleHomma, Keiichi, Hiroto Anbo, Tamotsu Noguchi, and Satoshi Fukuchi. 2018. "Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins" International Journal of Molecular Sciences 19, no. 12: 3860. https://doi.org/10.3390/ijms19123860
APA StyleHomma, K., Anbo, H., Noguchi, T., & Fukuchi, S. (2018). Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins. International Journal of Molecular Sciences, 19(12), 3860. https://doi.org/10.3390/ijms19123860