Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease


Abstract
1. AML Classifications
2. Genetic Landscape of AML
3. Association of genetic lesions in AML
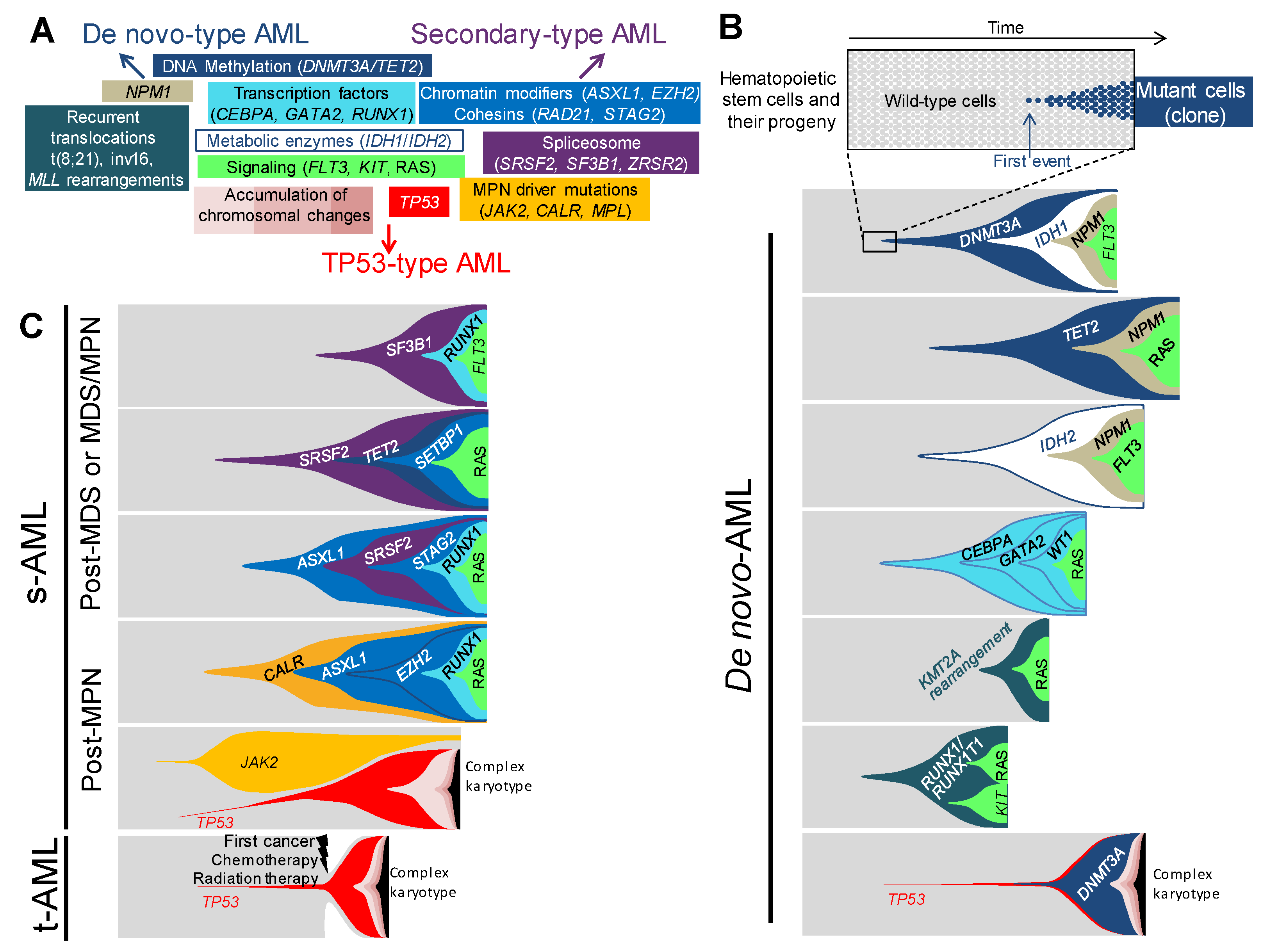
4. Distinct Genetic Hierarchies of AMLs
4.1. De novo-Type AMLs
4.2. Secondary-Type AMLs
4.2.1. Secondary-Type AMLs to Myelodysplastic Syndromes
4.2.2. Secondary-Type AMLs to Myeloproliferative Neoplasms
4.3. TP53-Type AMLs
4.4. Clonal Hierarchies in Children AML and Inherited AML Predisposition Syndromes
5. From Genetic Hierarchies to Clone-Specific Measurable Residual Disease in AML
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef]
- Hirsch, P.; Zhang, Y.; Tang, R.; Joulin, V.; Boutroux, H.; Pronier, E.; Moatti, H.; Flandrin, P.; Marzac, C.; Bories, D.; et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat. Commun. 2016, 7, 12475. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Tan, Y.; Lin, H.; Liu, X.; Yu, L.; Yang, Y.; Liu, S.; Bai, O.; Yang, Y.; Jin, F.; et al. Mutational spectrum of acute myeloid leukemia patients with double CEBPA mutations based on next-generation sequencing and its prognostic significance. Oncotarget 2018, 9, 24970–24979. [Google Scholar] [CrossRef] [PubMed]
- Avellino, R.; Delwel, R. Expression and regulation of C/EBPα in normal myelopoiesis and in malignant transformation. Blood 2017, 129, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Van Oevelen, C.; Collombet, S.; Vicent, G.; Hoogenkamp, M.; Lepoivre, C.; Badeaux, A.; Bussmann, L.; Sardina, J.L.; Thieffry, D.; Beato, M.; et al. C/EBPα activates pre-existing and de novo macrophage enhancers during induced pre-B cell transdifferentiation and myelopoiesis. Stem Cell Rep. 2015, 5, 232–247. [Google Scholar] [CrossRef]
- Hasemann, M.S.; Lauridsen, F.K.B.; Waage, J.; Jakobsen, J.S.; Frank, A.-K.; Schuster, M.B.; Rapin, N.; Bagger, F.O.; Hoppe, P.S.; Schroeder, T.; et al. C/EBPα is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014, 10, e1004079. [Google Scholar] [CrossRef]
- Wölfler, A.; Danen-van Oorschot, A.A.; Haanstra, J.R.; Valkhof, M.; Bodner, C.; Vroegindeweij, E.; van Strien, P.; Novak, A.; Cupedo, T.; Touw, I.P. Lineage-instructive function of C/EBPα in multipotent hematopoietic cells and early thymic progenitors. Blood 2010, 116, 4116–4125. [Google Scholar] [CrossRef] [PubMed]
- Fasan, A.; Haferlach, C.; Alpermann, T.; Jeromin, S.; Grossmann, V.; Eder, C.; Weissmann, S.; Dicker, F.; Kohlmann, A.; Schindela, S.; et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014, 28, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Sarojam, S.; Raveendran, S.; Vijay, S.; Sreedharan, J.; Narayanan, G.; Sreedharan, H. Characterization of CEBPA Mutations and Polymorphisms and their Prognostic relevance in de novo acute myeloid leukemia patients. Asian Pac. J. Cancer Prev. 2015, 16, 3785–3792. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Morgan, E.; Moodie, E.E.M.; Pullarkat, S.; Yeung, C.; Broesby-Olsen, S.; Ohgami, R.; Kim, Y.; Sperr, W.; Vestergaard, H.; et al. Core-binding factor acute myeloid leukemia with t(8;21): Risk factors and a novel scoring system (I-CBFit). Cancer Med. 2018, 7, 4447–4455. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Suzuki, M.; Watanabe, Y.; Takahashi, T.; Aoki, Y.; Uchiyama, T.; Kumaki, S.; Sasahara, Y.; Minegishi, M.; Kure, S.; et al. Development of a multi-step leukemogenesis model of MLL-rearranged leukemia using humanized mice. PLoS ONE 2012, 7, e37892. [Google Scholar] [CrossRef] [PubMed]
- Bäsecke, J.; Schwieger, M.; Griesinger, F.; Schiedlmeier, B.; Wulf, G.; Trümper, L.; Stocking, C. AML1/ETO promotes the maintenance of early hematopoietic progenitors in NOD/SCID mice but does not abrogate their lineage specific differentiation. Leuk. Lymphoma 2005, 46, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.; Krejci, O.; Wei, J.; Mulloy, J.C. Human CD34+ cells expressing the inv(16) fusion protein exhibit a myelomonocytic phenotype with greatly enhanced proliferative ability. Blood 2006, 108, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; de Albuquerque, A.; Nadarajah, N.; Alpermann, T.; Kern, W.; Steuer, K.; Perglerová, K.; Haferlach, C.; Schnittger, S.; Haferlach, T. Karyotype evolution and acquisition of FLT3 or RAS pathway alterations drive progression of myelodysplastic syndrome to acute myeloid leukemia. Haematologica 2015, 100, e487–e490. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Chesnais, V.; Arcangeli, M.L.; Delette, C.; Rousseau, A.; Guermouche, H.; Lefevre, C.; Bondu, S.; Cheok, M.; Chapuis, N.; Legros, L.; et al. Architectural and functional heterogeneity of hematopoietic stem/progenitor cells in non-del(5q) myelodysplastic syndromes. Blood 2017, 129, 484–496. [Google Scholar] [CrossRef]
- Chesnais, V.; Renneville, A.; Toma, A.; Lambert, J.; Passet, M.; Dumont, F.; Chevret, S.; Lejeune, J.; Raimbault, A.; Stamatoullas, A.; et al. Effect of lenalidomide treatment on clonal architecture of myelodysplastic syndromes without 5q deletion. Blood 2016, 127, 749–760. [Google Scholar] [CrossRef]
- da Silva-Coelho, P.; Kroeze, L.I.; Yoshida, K.; Koorenhof-Scheele, T.N.; Knops, R.; van de Locht, L.T.; de Graaf, A.O.; Massop, M.; Sandmann, S.; Dugas, M.; et al. Clonal evolution in myelodysplastic syndromes. Nat. Commun. 2017, 8, 15099. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Shao, J.; Ding, L.; White, B.S.; Kandoth, C.; Miller, C.A.; Niu, B.; McLellan, M.D.; Dees, N.D.; et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 2013, 27, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Mossner, M.; Jann, J.-C.; Wittig, J.; Nolte, F.; Fey, S.; Nowak, V.; Obländer, J.; Pressler, J.; Palme, I.; Xanthopoulos, C.; et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood 2016, 128, 1246–1259. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627, quiz 3699. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Morabito, M.; Preudhomme, C.; Berthon, C.; Adès, L.; Fenaux, P.; Platzbecker, U.; Gagey, O.; et al. Clonal architecture of chronic myelomonocytic leukemias. Blood 2013, 121, 2186–2198. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-L.; Nagata, Y.; Kao, H.-W.; Sanada, M.; Okuno, Y.; Huang, C.-F.; Liang, D.-C.; Kuo, M.-C.; Lai, C.-L.; Lee, E.-H.; et al. Clonal leukemic evolution in myelodysplastic syndromes with TET2 and IDH1/2 mutations. Haematologica 2014, 99, 28–36. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513, quiz 2615. [Google Scholar] [CrossRef] [PubMed]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.G.; Ortmann, C.A.; Green, A.R. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 1865–1866. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Fasan, A.; Haferlach, C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: An analysis of 3307 cases. Leukemia 2017, 31, 705–711. [Google Scholar] [CrossRef]
- Wong, T.N.; Miller, C.A.; Klco, J.M.; Petti, A.; Demeter, R.; Helton, N.M.; Li, T.; Fulton, R.S.; Heath, S.E.; Mardis, E.R.; et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016, 127, 893–897. [Google Scholar] [CrossRef]
- Granfeldt Østgård, L.S.; Medeiros, B.C.; Sengeløv, H.; Nørgaard, M.; Andersen, M.K.; Dufva, I.H.; Friis, L.S.; Kjeldsen, E.; Marcher, C.W.; Preiss, B.; et al. Epidemiology and Clinical Significance of Secondary and Therapy-Related Acute Myeloid Leukemia: A National Population-Based Cohort Study. J. Clin. Oncol. 2015, 33, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Marceau-Renaut, A.; Duployez, N.; Ducourneau, B.; Labopin, M.; Petit, A.; Rousseau, A.; Geffroy, S.; Bucci, M.; Cuccuini, W.; Fenneteau, O.; et al. Molecular profiling defines distinct prognostic subgroups in childhood AML: A Report from the French ELAM02 study group. HemaSphere 2018, 2, e31. [Google Scholar] [CrossRef]
- Creutzig, U.; Zimmermann, M.; Reinhardt, D.; Rasche, M.; von Neuhoff, C.; Alpermann, T.; Dworzak, M.; Perglerová, K.; Zemanova, Z.; Tchinda, J.; et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer 2016, 122, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Gale, K.B.; Ford, A.M.; Repp, R.; Borkhardt, A.; Keller, C.; Eden, O.B.; Greaves, M.F. Backtracking leukemia to birth: Identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl. Acad. Sci. USA 1997, 94, 13950–13954. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014, 51, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.J.; Kuiper, R.P.; Carmichael, C.L.; Wilkins, E.J.; Dors, N.; Carmagnac, A.; Schouten-van Meeteren, A.Y.N.; Li, X.; Stankovic, M.; Kamping, E.; et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: Clues for improved identification of the FPD/AML syndrome. Leukemia 2010, 24, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Donadieu, J.; Fenneteau, O.; Beaupain, B.; Beaufils, S.; Bellanger, F.; Mahlaoui, N.; Lambilliotte, A.; Aladjidi, N.; Bertrand, Y.; Mialou, V.; et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica 2012, 97, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E.; et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef]
- Donadieu, J.; Lamant, M.; Fieschi, C.; de Fontbrune, F.S.; Caye, A.; Ouachee, M.; Beaupain, B.; Bustamante, J.; Poirel, H.A.; Isidor, B.; et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 2018, 103, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Cavenagh, J.D.; Lister, T.A.; Fitzgibbon, J. Mutation of CEBPA in Familial Acute Myeloid Leukemia. N. Engl. J. Med. 2004, 351, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, M.; Walne, A.J.; Plagnol, V.; Velangi, M.; Ho, A.; Hossain, U.; Vulliamy, T.; Dokal, I. Exome Sequencing Identifies Autosomal-Dominant SRP72 Mutations Associated with Familial Aplasia and Myelodysplasia. Am. J. Hum. Genet. 2012, 90, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Saliba, J.; Saint-Martin, C.; di Stefano, A.; Lenglet, G.; Marty, C.; Keren, B.; Pasquier, F.; Valle, V.D.; Secardin, L.; Leroy, G.; et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 2015, 47, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef]
- Link, D.C.; Schuettpelz, L.G.; Shen, D.; Wang, J.; Walter, M.J.; Kulkarni, S.; Payton, J.E.; Ivanovich, J.; Goodfellow, P.J.; Beau, M.L.; et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA 2011, 305, 1568–1576. [Google Scholar] [CrossRef]
- Xia, J.; Miller, C.A.; Baty, J.; Ramesh, A.; Jotte, M.R.M.; Fulton, R.S.; Vogel, T.P.; Cooper, M.A.; Walkovich, K.J.; Makaryan, V.; et al. Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood 2018, 131, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Donadieu, J.; Delhommeau, F. TP53 mutations: The dawn of Shwachman clones. Blood 2018, 131, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- San Miguel, J.F.; Vidriales, M.B.; López-Berges, C.; Díaz-Mediavilla, J.; Gutiérrez, N.; Cañizo, C.; Ramos, F.; Calmuntia, M.J.; Pérez, J.J.; González, M.; et al. Early immunophenotypical evaluation of minimal residual disease in acute myeloid leukemia identifies different patient risk groups and may contribute to postinduction treatment stratification. Blood 2001, 98, 1746–1751. [Google Scholar] [CrossRef]
- Venditti, A.; Buccisano, F.; del Poeta, G.; Maurillo, L.; Tamburini, A.; Cox, C.; Battaglia, A.; Catalano, G.; Del Moro, B.; Cudillo, L.; et al. Level of minimal residual disease after consolidation therapy predicts outcome in acute myeloid leukemia. Blood 2000, 96, 3948–3952. [Google Scholar]
- Terwijn, M.; van Putten, W.L.J.; Kelder, A.; van der Velden, V.H.J.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; de Greef, G.E.; Valk, P.J.M.; et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: Data from the HOVON/SAKK AML 42A study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef]
- Freeman, S.D.; Virgo, P.; Couzens, S.; Grimwade, D.; Russell, N.; Hills, R.K.; Burnett, A.K. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J. Clin. Oncol. 2013, 31, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- MRD-AML-BFM Study Group; Langebrake, C.; Creutzig, U.; Dworzak, M.; Hrusak, O.; Mejstrikova, E.; Griesinger, F.; Zimmermann, M.; Reinhardt, D. Residual disease monitoring in childhood acute myeloid leukemia by multiparameter flow cytometry: The MRD-AML-BFM Study Group. J. Clin. Oncol. 2006, 24, 3686–3692. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.A.L.; O’Brien, M.A.; Hills, R.K.; Daly, S.B.; Wheatley, K.; Burnett, A.K. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: Results of the United Kingdom MRC AML-15 trial. Blood 2012, 120, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Liu Yin, J.A. Minimal residual disease in acute myeloid leukaemia. Best Pract. Res. Clin. Haematol. 2002, 15, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Willekens, C.; Blanchet, O.; Renneville, A.; Cornillet-Lefebvre, P.; Pautas, C.; Guieze, R.; Ifrah, N.; Dombret, H.; Jourdan, E.; Preudhomme, C.; et al. Prospective long-term minimal residual disease monitoring using RQ-PCR in RUNX1-RUNX1T1-positive acute myeloid leukemia: Results of the French CBF-2006 trial. Haematologica 2016, 101, 328–335. [Google Scholar] [CrossRef]
- Grimwade, D.; Jovanovic, J.V.; Hills, R.K.; Nugent, E.A.; Patel, Y.; Flora, R.; Diverio, D.; Jones, K.; Aslett, H.; Batson, E.; et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J. Clin. Oncol. 2009, 27, 3650–3658. [Google Scholar] [CrossRef]
- Platzbecker, U.; Avvisati, G.; Cicconi, L.; Thiede, C.; Paoloni, F.; Vignetti, M.; Ferrara, F.; Divona, M.; Albano, F.; Efficace, F.; et al. Improved Outcomes with Retinoic Acid and Arsenic Trioxide Compared with Retinoic Acid and Chemotherapy in Non-High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J. Clin. Oncol. 2017, 35, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Schlenk, R.F.; Jensen, K.-O.; Tschürtz, F.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Onken, S.; Eiwen, K.; Habdank, M.; et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: A study from the German-Austrian acute myeloid leukemia study group. J. Clin. Oncol. 2011, 29, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Shayegi, N.; Kramer, M.; Bornhäuser, M.; Schaich, M.; Schetelig, J.; Platzbecker, U.; Röllig, C.; Heiderich, C.; Landt, O.; Ehninger, G.; et al. The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood 2013, 122, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Balsat, M.; Renneville, A.; Thomas, X.; de Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.-P.; Nibourel, O.; Berthon, C.; et al. Postinduction Minimal Residual Disease Predicts Outcome and Benefit From Allogeneic Stem Cell Transplantation in Acute Myeloid Leukemia With NPM1 Mutation: A Study by the Acute Leukemia French Association Group. J. Clin. Oncol. 2017, 35, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: A European LeukemiaNet study. J. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef] [PubMed]
- Nomdedéu, J.F.; Hoyos, M.; Carricondo, M.; Bussaglia, E.; Estivill, C.; Esteve, J.; Tormo, M.; Duarte, R.; Salamero, O.; de Llano, M.P.Q.; et al. Bone marrow WT1 levels at diagnosis, post-induction and post-intensification in adult de novo AML. Leukemia 2013, 27, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015, 314, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X.; et al. Clearance of Somatic Mutations at Remission and the Risk of Relapse in Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Gaksch, L.; Kashofer, K.; Heitzer, E.; Quehenberger, F.; Daga, S.; Hofer, S.; Halbwedl, I.; Graf, R.; Krisper, N.; Hoefler, G.; et al. Residual disease detection using targeted parallel sequencing predicts relapse in cytogenetically normal acute myeloid leukemia. Am. J. Hematol. 2018, 93, 23–30. [Google Scholar] [CrossRef]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Nibourel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the Acute Leukemia French Association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [CrossRef]
- Bhatnagar, B.; Eisfeld, A.-K.; Nicolet, D.; Mrózek, K.; Blachly, J.S.; Orwick, S.; Lucas, D.M.; Kohlschmidt, J.; Blum, W.; Kolitz, J.E.; et al. Persistence of DNMT3A R882 mutations during remission does not adversely affect outcomes of patients with acute myeloid leukaemia. Br. J. Haematol. 2016, 175, 226–236. [Google Scholar] [CrossRef]
- Hirsch, P.; Tang, R.; Abermil, N.; Flandrin, P.; Moatti, H.; Favale, F.; Suner, L.; Lorre, F.; Marzac, C.; Fava, F.; et al. Precision and prognostic value of clone-specific minimal residual disease in acute myeloid leukemia. Haematologica 2017, 102, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Londoño-Joshi, A.; Kang, Q.; Tewari, M.; Rhim, A.D.; Malek, S.N. Ultrasensitive mutation detection identifies rare residual cells causing acute myelogenous leukemia relapse. J. Clin. Investig. 2017, 127, 3484–3495. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg-Thurley, M.; Amler, S.; Goerlich, D.; Köhnke, T.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Herold, T.; Hubmann, M.; Ksienzyk, B.; et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M.; et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Young, A.L.; Wong, T.N.; Hughes, A.E.O.; Heath, S.E.; Ley, T.J.; Link, D.C.; Druley, T.E. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia 2015, 29, 1608–1611. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Ueno, T.; Fukumura, K.; Yamato, A.; Ando, M.; Yamaguchi, H.; Soda, M.; Kawazu, M.; Sai, E.; Yamashita, Y.; et al. Leukemic evolution of donor-derived cells harboring IDH2 and DNMT3A mutations after allogeneic stem cell transplantation. Leukemia 2014, 28, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, P.; Mamez, A.C.; Belhocine, R.; Lapusan, S.; Tang, R.; Suner, L.; Bories, D.; Marzac, C.; Fava, F.; Legrand, O.; et al. Clonal history of a cord blood donor cell leukemia with prenatal somatic JAK2 V617F mutation. Leukemia 2016, 30, 1756–1759. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Category of lesions | Mutated Gene or Cytogenetic Lesion | Encoded Protein | European Leukemia Net Prognostic Group | Pairwise Exclusion of Lesions * | Pairwise Association of Lesions * |
|---|---|---|---|---|---|
| DNA methylation | DNMT3A | DNA Methyltransferase 3α | NA | CEBPA(monoallelic), complex karyotype, KIT, ASXL1, SRSF2, del(5q)/−5, recurrent translocations | NPM1, FLT3, IDH1, IDH2, PTPN11 |
| TET2 | Tet Methylcytosine Dioxygenase 2 | NA | IDH2, ASXL1, SRSF2, complex karyotype, del(5q)/−5, del(7q)/−7, t(15;17) | NPM1, SRSF2, STAG2, FLT3 | |
| Metabolic enzymes | IDH1 | Isocitrate Dehydrogenase (NADP(+)) 1, Cytosolic | NA | TP53, recurrent translocations, complex karyotype | DNMT3A, NPM1, PTPN11 |
| IDH2 | Isocitrate Dehydrogenase (NADP(+)) 2, Mitochondrial | NA | TP53, NRAS, FLT3-ITD, recurrent translocations, complex karyotype | DNMT3A, SRSF2, NPM1 | |
| Nucleophosmin 1 | NPM1 | Nucleophosmin 1 | Favorable (without FLT3-ITD) | Abnormal karyotype, TP53, CEBPA (bi-allelic), RUNX1, U2AF1, ASXL1, SF3B1, EZH2, KIT | DNMT3A, TET2, IDH1, RAD21, FLT3, PTPN11, IDH2 |
| Transcription factors | CEBPA | CCAAT Enhancer Binding Protein α | Favorable (bi-allelic mutation) | NPM1, DNMT3A, RUNX1, NRAS (if CEBPA mono-allelic) complex karyotype, del(5q)/−5, | GATA2, WT1, STAG2 |
| ETV6 | ETS Variant 6 | NA | None | SF3B1, KRAS, inv(3) | |
| GATA2 | GATA Binding Protein 2 | NA | None | CEBPA, NRAS | |
| RUNX1 | Runt Related Transcription Factor 1 | Adverse | NPM1, CEBPA (bi-allelic), TP53, recurrent translocations | ASXL1, SRSF2, EZH2, PHF6, STAG2, BCOR | |
| WT1 | Wilms Tumor 1 | NA | DNMT3A, CEBPA (mono-allelic), TP53, complex karyotype, del(5q)/−5 | CEBPA, FLT3, t(15;17) | |
| Signaling | CBL | Cbl Proto-Oncogene | NA | None | SF3B1 |
| FLT3 | Fms Related Tyrosine Kinase 3 | Adverse (if ITD, NPM1 wild type, and high allelic ratio) | TP53, NRAS, inv(16), del(17p)/−17, del(5q)/−5, del(7q)/−7, complex karyotype, KRAS, NF1, KIT, SRSF2, ASXL1, del(20q)/−20, inv(3) | DNMT3A, NPM1, t(6;9), t(15;17), WT1 | |
| KIT | KIT Proto-Oncogene Receptor Tyrosine Kinase | NA | NPM1, DNMT3A, FLT3-ITD | t(8;21), inv(16) | |
| KRAS | KRAS Proto-Oncogene, GTPase | NA | FLT3-ITD | ETV6, inv(3) | |
| NF1 | Neurofibromin 1 | NA | FLT3-ITD | None | |
| NRAS | NRAS Proto-Oncogene, GTPase | NA | FLT3-ITD, t(15;17), IDH2, CEBPA (mono-allelic), TP53, complex karyotype | inv(16), GATA2 | |
| PTPN11 | Protein Tyrosine Phosphatase, Non-Receptor Type 11 | NA | t(15;17), t(8;21) | NPM1, DNMT3A, IDH1, inv(3), del(7q)/-7 | |
| Chromatin/Cohesin | PHF6 | PHD Finger Protein 6 | NA | None | RUNX1 |
| ASXL1 | Aditionnal Sex Comb Like 1 | Adverse | FLT3-ITD | RUNX1, SRSF2, STAG2, EZH2, U2AF1 | |
| BCOR | BCL6 Corepressor | NA | FLT3-ITD | SRSF2, RUNX1, inv(3) | |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit | NA | None | RUNX1, ASXL1, STAG2 | |
| RAD21 | RAD21 Cohesin Complex Component | NA | None | NPM1, t(8;21) | |
| STAG2 | Stromal Antigen 2 | NA | None | SRSF2, ASXL1, BCOR, RUNX1, TET2, EZH2, CEBPA | |
| Spliceosome | SF3B1 | Splicing Factor 3b Subunit 1 | NA | NPM1 | ETV6, CBL, inv(3) |
| SRSF2 | Serine And Arginine Rich Splicing Factor 2 | NA | DNMT3A, inv(16) | ASXL1, RUNX1, STAG2, TET2, IDH2, BCOR | |
| U2AF1 | U2 Small Nuclear RNA Auxiliary Factor 1 | NA | NPM1 | ASXL1, del(20q)/-20 | |
| Tumor suppressor | TP53 | Tumor Protein P53 | Adverse | NPM1, FLT3, NRAS, WT1, IDH1, IDH2, RUNX1 | Complex karyotype, del(5q)/−5, del(7q)/−7, del(20q)/−20 |
| Gene fusions | t(8;21) | RUNX1-RUNX1T1 | Favorable | NPM1, DNMT3A, IDH2, RUNX1, FLT3-ITD, PTPN11, complex karyotype | KIT, RAD21 |
| t(15;17) | PML-RARA | Favorable | NPM1, DNMT3A, TET2, IDH2, RUNX1, PTPN11, complex karyotype | WT1, FLT3 | |
| inv(16)/t(16;16) | CBFB-MYH11 | Favorable | NPM1, DNMT3A, IDH1, IDH2, RUNX1, SRSF2, complex karyotype, del(5q)/−5 | NRAS, KIT | |
| t(6;9) | DEK-NUP214 | Adverse | NPM1 | FLT3 | |
| inv(3) | RPN1-MECOM | Adverse | NPM1, FLT3-ITD | del(7q)/−7, ETV6, SF3B1, BCOR, KRAS, PTPN11 | |
| t(9;11) | MLLT3-KMT2A | Intermediate | NPM1, DNMT3A | None | |
| Complex karyotype | Complex karyotype | NA | Adverse | NPM1, DNMT3A, FLT3, recurrent translocations, NRAS, WT1, TET2, IDH1, IDH2 | TP53 |
| Other cytogenetic lesions | del(5q) or −5 | NA | Adverse | NPM1, FLT3, DNMT3A, TET2, IDH2, WT1, CEBPA (bi-allelic), inv(16) | TP53 |
| del(7q) or −7 | NA | Adverse | NPM1, FLT3-ITD, DNMT3A, TET2, WT1 | TP53, inv(3), PTPN11 | |
| abn(17p) or −17 | NA | Adverse | NPM1, FLT3-ITD, DNMT3A, TET2 | TP53 | |
| del(20q) or −20 | NA | Intermediate | NPM1, FLT3-ITD | TP53, U2AF1 | |
| other KMT2A translocations | NA | Adverse (except t(9;11)) | NPM1 | None |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martignoles, J.-A.; Delhommeau, F.; Hirsch, P. Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. Int. J. Mol. Sci. 2018, 19, 3850. https://doi.org/10.3390/ijms19123850
Martignoles J-A, Delhommeau F, Hirsch P. Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. International Journal of Molecular Sciences. 2018; 19(12):3850. https://doi.org/10.3390/ijms19123850
Chicago/Turabian StyleMartignoles, Jean-Alain, François Delhommeau, and Pierre Hirsch. 2018. "Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease" International Journal of Molecular Sciences 19, no. 12: 3850. https://doi.org/10.3390/ijms19123850
APA StyleMartignoles, J.-A., Delhommeau, F., & Hirsch, P. (2018). Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. International Journal of Molecular Sciences, 19(12), 3850. https://doi.org/10.3390/ijms19123850