Unambiguous Ex Situ and in Cell 2D 13C Solid-State NMR Characterization of Starch and Its Constituents

Abstract
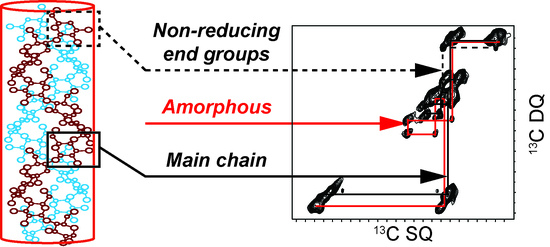
1. Introduction
2. Results and Discussion
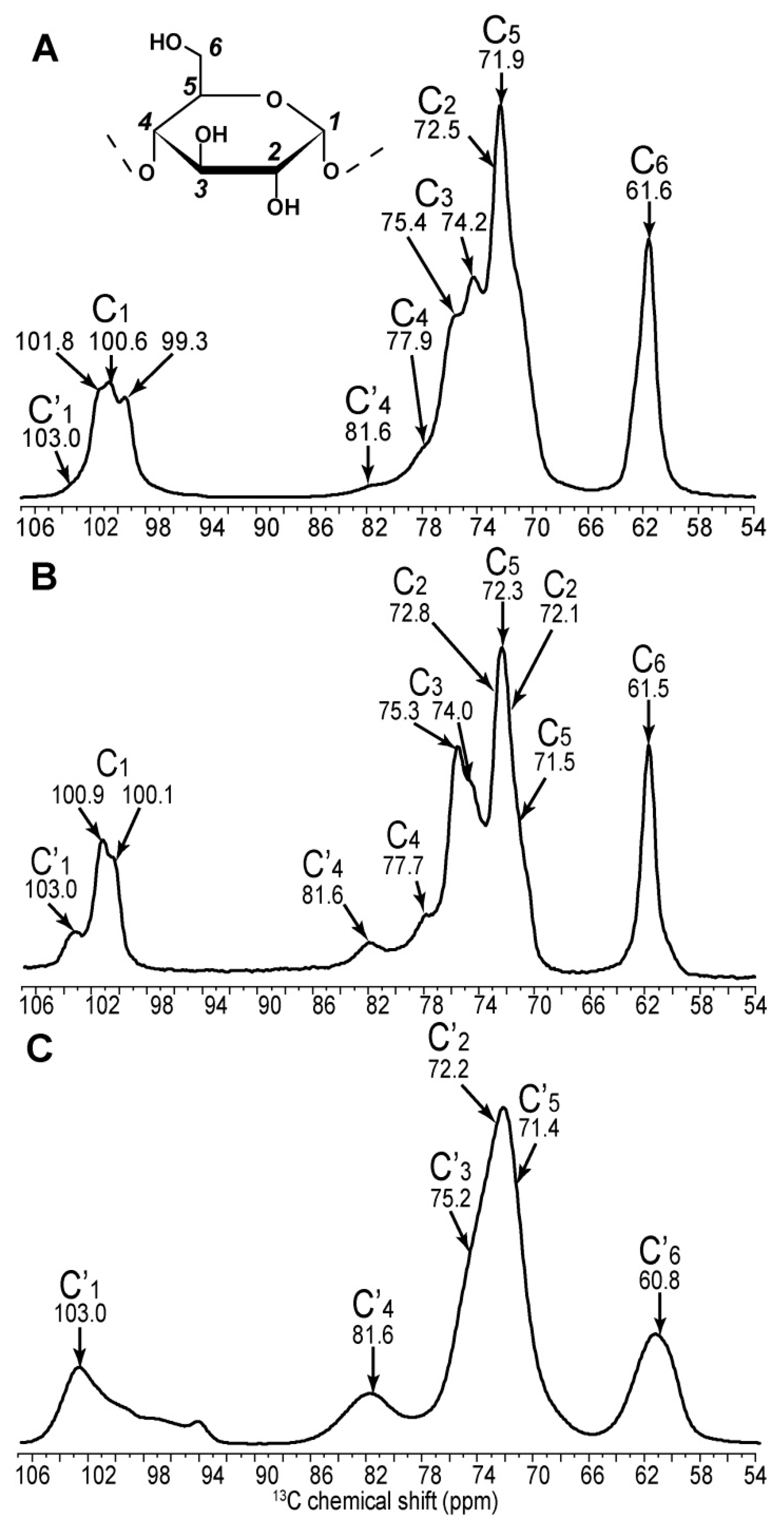
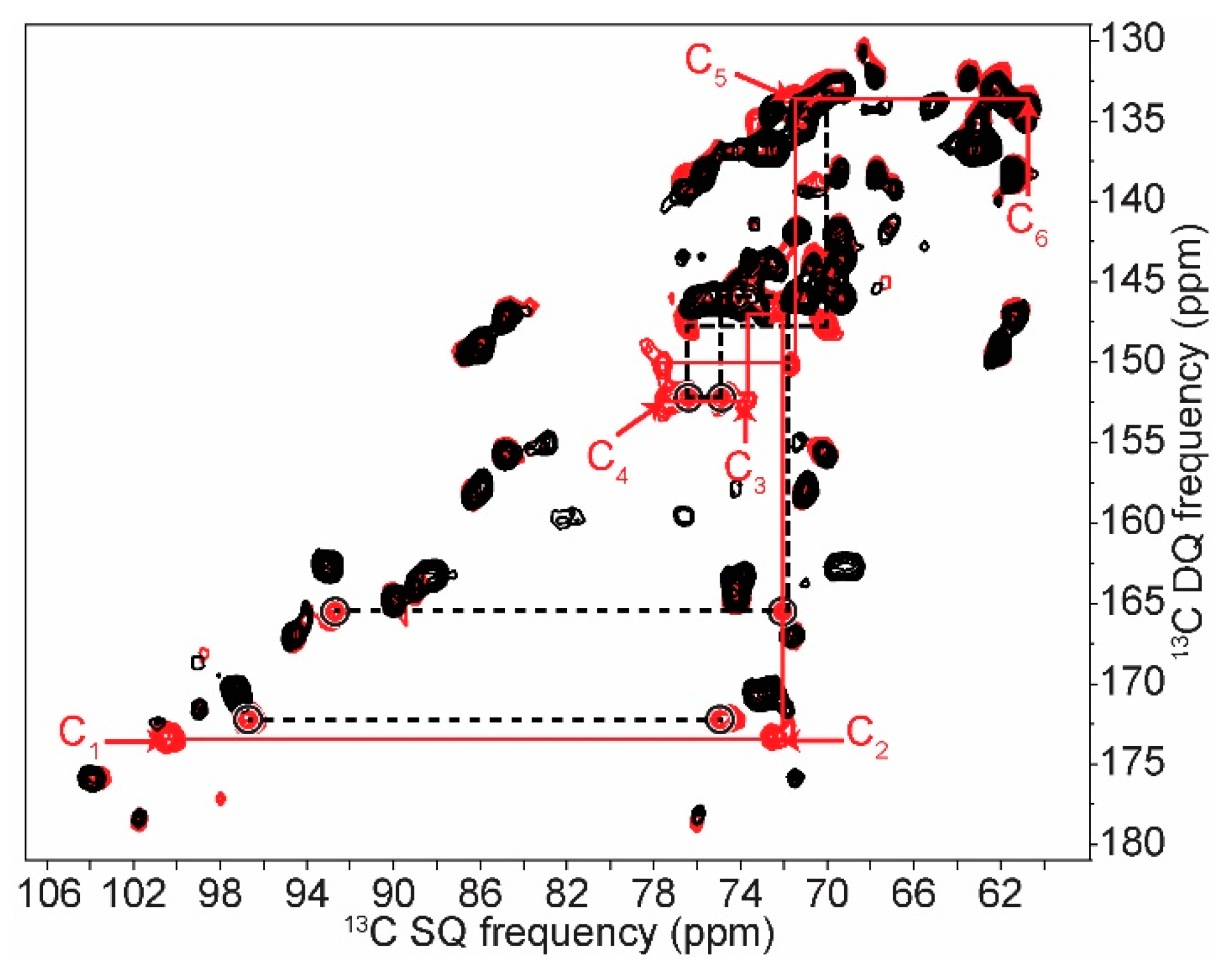
2.1. 2D 13C SS-NMR Ex Situ Characterization of A and B Types and Amorphous Starch
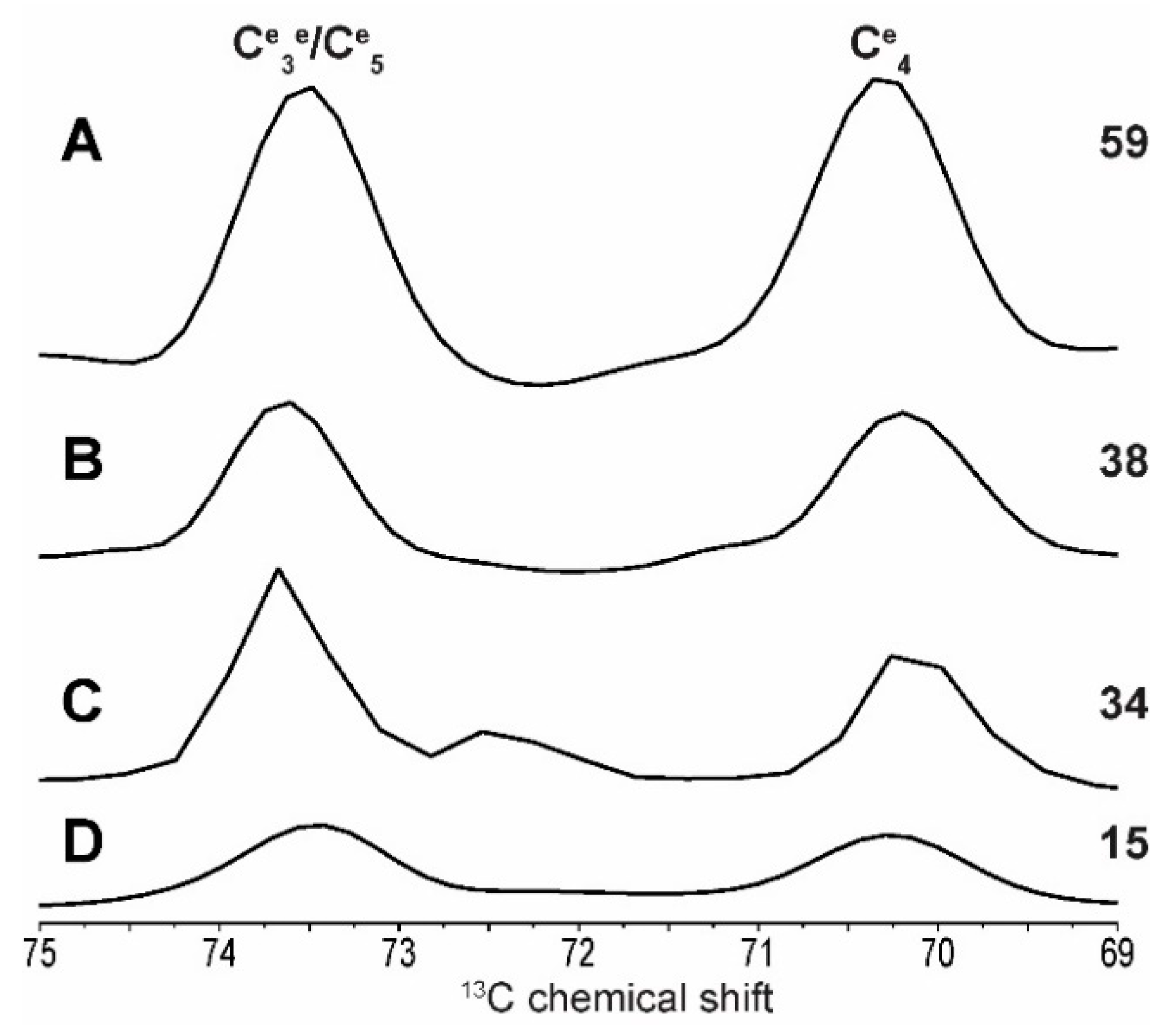
2.2. Differentiating Between Starch Components: Amylose and Amylopectin
2.3. In Cell Characterization of C. reinhardtii Starch
3. Materials and Methods
3.1. Materials
3.2. Strain, Media and Growth Conditions
3.3. Starch Purification
3.4. Starch Retrogradation
3.5. Cell Viability
3.6. Solid-State NMR
3.7. X-ray Diffraction
3.8. Crystallinity Quantification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two dimensional |
| SS-NMR | Solid state Nuclear Magnetic Resonance |
| 13C | Carbon 13 |
| µm | Micrometer |
| nm | Nanometer |
| XRD | X-ray diffraction |
| 1D | One dimensional |
| g/L | Gram per liter |
| rpm | Rotation per minute |
| NaH13CO3 | Sodium bicarbonate |
| mg | Milligram |
| mm | Millimeter |
| mL | Milliliter |
| mM | Millimole per liter |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| EDTA | Ethylenediaminetetraacetic acid |
| PMSF | Phenylmethylsulfonyl fluoride |
| FDA | Fluorescein DiAcetate |
| MHz | Mega Hertz |
| MAS | Magic Angle Spinning |
| 1H | Proton |
| kHz | Kilo Hertz |
| SP | Single pulse |
| CP | Cross polarization |
| INADEQUATE | Incredible Natural Abundance Double QUAntum Technique |
| µs | Microseconde |
| TPPM | Two-pulse phase modulation |
| NOE | Nuclear Overhauser Effect |
| TPPI | Time Proportional Phase Incrementation |
| Å | Ångström |
| DQ | Double quantum |
| ppm | Parts per million |
| wt | Wild type |
| ap | Amylopectin-rich starch |
| as | Amylose-rich starch |
| retro | Retrograded wild-type starch |
| am | Amorphuous starch |
References
- Pérez, S.; Bertoft, E. The molecular structures of starch components and their contribution to the architecture of starch granules: A comprehensive review. Starch/Staerke 2010, 62, 389–420. [Google Scholar] [CrossRef]
- Sajilata, M.G.; Singhal, R.S.; Kulkarni, P.R. Resistant starch—A review. Compr. Rev. Food Sci. Food Saf. 2006, 5, 1–17. [Google Scholar] [CrossRef]
- Paleos, C.M.; Sideratou, Z.; Tsiourvas, D. Drug delivery systems based on hydroxyethyl starch. Bioconjug. Chem. 2017, 28, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Bertoft, E. Understanding starch structure: Recent progress. Agronomy 2017, 7, 56. [Google Scholar] [CrossRef]
- Sarko, A.; Zugenmaier, P. Crystal structures of amylose and its derivatives. In Fiber Diffraction Methods; American Chemical Society: Washington, DC, USA, 1980; Volume 141, pp. 459–482. [Google Scholar]
- Wu Hsien-Chih, H.; Sarko, A. The double-helical molecular structure of crystalline A-amylose. Carbohydr. Res. 1978, 61, 27–40. [Google Scholar] [CrossRef]
- Wu Hsein-Chih, H.; Sarko, A. The double-helical molecular structure of crystalline B-amylose. Carbohydr. Res. 1978, 61, 7–25. [Google Scholar] [CrossRef]
- Popov, D.; Buléon, A.; Burghammer, M.; Chanzy, H.; Montesanti, N.; Putaux, J.L.; Potocki-Véronèse, G.; Riekel, C. Crystal structure of A-amylose: A revisit from synchrotron microdiffraction analysis of single crystals. Macromolecules 2009, 42, 1167–1174. [Google Scholar] [CrossRef]
- Imberty, A.; Chanzy, H.; Perez, S.; Buleon, A.; Tran, V. The double-helical nature of the crystalline part of A-starch. J. Mol. Biol. 1988, 201, 365–378. [Google Scholar] [CrossRef]
- Imberty, A.; Perez, S. A revisit to the three-dimensional structure of B-type starch. Biopolymers 1988, 27, 1205–1221. [Google Scholar] [CrossRef]
- Buleon, A.; Gallant, D.J.; Bouchet, B.; Mouille, G.; D’Hulst, C.; Kossmann, J.; Ball, S. Starches from A to C (Chlamydomonas reinhardtii as a Model Microbial System to Investigate the Biosynthesis of the Plant Amylopectin Crystal). Plant Physiol. 1997, 115, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Rondeau-Mouro, C.; Buléon, A.; Lahaye, M. Caractérisation par RMN des biopolymères d’origine végétale, de la molécule à l’organisation supramoléculaire. C. R. Chim. 2008, 11, 370–379. [Google Scholar] [CrossRef]
- Paris, M.B.H.; Emery, J.; Buzaré, J.Y.; Buléon, A. Crystallinity and structuring role of water in native and recrystallized starches by 13C CP-MAS NMR spectroscopy: 1: Spectral decomposition. Carbohydr. Polym. 1999, 39, 13. [Google Scholar] [CrossRef]
- Rondeau-Mouro, C.; Veronese, G.; Buleon, A. High-resolution solid-state NMR of B-type amylose. Biomacromolecules 2006, 7, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Paris, M.; Bizot, H.; Emery, J.; Buzare, J.Y.; Buleon, A. NMR local range investigations in amorphous starchy substrates I. Structural heterogeneity probed by (13)C CP-MAS NMR. Int. J. Biol. Macromol. 2001, 29, 127–136. [Google Scholar] [CrossRef]
- Paris, M.; Bizot, H.; Emery, J.; Buzare, J.Y.; Buleon, A. NMR local range investigations in amorphous starchy substrates: II-Dynamical heterogeneity probed by (1)H/(13)C magnetization transfer and 2D WISE solid state NMR. Int. J. Biol. Macromol. 2001, 29, 137–143. [Google Scholar] [CrossRef]
- Crow, W.B. Chapter IV—The cell. In A Synopsis of Biology; Crow, W.B., Ed.; Butterworth-Heinemann: Oxford, UK, 1960; pp. 10–24. [Google Scholar]
- Johnson, X.; Alric, J. Central carbon metabolism and electron transport in Chlamydomonas reinhardtii: Metabolic constraints for carbon partitioning between oil and starch. Eukaryot. Cell 2013, 12, 776–793. [Google Scholar] [CrossRef] [PubMed]
- Rolland, N.; Atteia, A.; Decottignies, P.; Garin, J.; Hippler, M.; Kreimer, G.; Lemaire, S.D.; Mittag, M.; Wagner, V. Chlamydomonas proteomics. Curr. Opin. Microbiol. 2009, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Li-Beisson, Y.; Beisson, F.; Riekhof, W. Metabolism of acyl-lipids in Chlamydomonas reinhardtii. Plant J. 2015, 82, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Blaby, I.K.; Blaby-Haas, C.; Tourasse, N.; Hom, E.F.Y.; Lopez, D.; Aksoy, M.; Grossman, A.; Umen, J.; Dutcher, S.; Porter, M.; et al. The Chlamydomonas genome project: A decade on. Trends Plant Sci. 2014, 19, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, S.D.; Fitz-Gibbon, S.T.; Glaesener, A.G.; Pellegrini, M.; Merchant, S.S. Chlamydomonas genome resource for laboratory strains reveals a mosaic of sequence variation, identifies true strain histories, and enables strain-specific studies. Plant Cell 2015, 27, 2335–2352. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.G.; Dirick, L.; Decq, A.; Martiat, J.-C.; Matagne, R. Physiology of starch storage in the monocellular alga Chlamydomonas reinhardtii. Plant Sci. 1990, 66, 1–9. [Google Scholar] [CrossRef]
- Gérard, C.; Colonna, P.; Buléon, A.; Planchot, V. Order in maize mutant starches revealed by mild acid hydrolysis. Carbohydr. Polym. 2002, 48, 131–141. [Google Scholar] [CrossRef]
- Perry, P.A.; Donald, A.M. The Role of Plasticization in Starch Granule Assembly. Biomacromolecules 2000, 1, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.R.; Godward, J.; Hills, B. The distribution of water in native starch granules—A multinuclear NMR study. Carbohydr. Polym. 2000, 43, 375–387. [Google Scholar] [CrossRef]
- Gidley, M.J.; Bociek, S.M. Molecular organization in starches: A carbon 13 CP/MAS NMR study. J. Am. Chem. Soc. 1985, 107, 7040–7044. [Google Scholar] [CrossRef]
- Arnold, A.A.; Bourgouin, J.P.; Genard, B.; Warschawski, D.E.; Tremblay, R.; Marcotte, I. Whole cell solid-state NMR study of Chlamydomonas reinhardtii microalgae. J. Biomol. NMR 2018, 70, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lesage, A.; Auger, C.; Caldarelli, S.; Emsley, L. Determination of Through-bond carbon–carbon connectivities in solid-state NMR using the inadequate experiment. J. Am. Chem. Soc. 1997, 119, 7867–7868. [Google Scholar] [CrossRef]
- Holland, G.P.; Jenkins, J.E.; Creager, M.S.; Lewis, R.V.; Yarger, J.L. Quantifying the fraction of glycine and alanine in β-sheet and helical conformations in spider dragline silk using solid-state NMR. Chem. Commun. 2008, 5568–5570. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.J.; Mortimer, J.C.; Bernardinelli, O.D.; Pöppler, A.-C.; Brown, S.P.; deAzevedo, E.R.; Dupree, R.; Dupree, P. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat. Commun. 2016, 7, 13902. [Google Scholar] [CrossRef] [PubMed]
- Idström, A.; Schantz, S.; Sundberg, J.; Chmelka, B.F.; Gatenholm, P.; Nordstierna, L. 13C NMR assignments of regenerated cellulose from solid-state 2D NMR spectroscopy. Carbohydr. Polym. 2016, 151, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Hong, M. Solid-state dipolar inadequate NMR spectroscopy with a large double-quantum spectral width. J. Magn. Reson. 1999, 136, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Warschawski, D.E.; Devaux, P.F. Polarization transfer in lipid membranes. J. Magn. Reson. 2000, 145, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.O.; Motawie, M.S.; Møller, B.L.; Hindsgaul, O.; Meier, S. NMR characterization of chemically synthesized branched α-dextrin model compounds. Carbohydr. Res. 2015, 403, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Jia, L.; Gao, J.; Song, Y.; Zhao, H.; Nakamura, Y.; Wu, D. The influences of chain length of amylopectin on resistant starch in rice (Oryza sativa L.). Starch/Staerke 2007, 59, 504–509. [Google Scholar] [CrossRef]
- Itoh, Y.; Crofts, N.; Abe, M.; Hosaka, Y.; Fujita, N. Characterization of the endosperm starch and the pleiotropic effects of biosynthetic enzymes on their properties in novel mutant rice lines with high resistant starch and amylose content. Plant Sci. 2017, 258, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.B.; Gangoda, M.; Gilpin, R.K.; Su, W. The influence of hydration on the conformation of lysozyme studied by solid-state 13C-nmr spectroscopy. Biopolymers 1993, 33, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Stypa, M.P.; Tenn, B.K.; Kumashiro, K.K. Solid-state (13)C NMR reveals effects of temperature and hydration on elastin. Biophys. J. 2002, 82, 1086–1095. [Google Scholar] [CrossRef]
- Sakellariou, D.; Brown, S.P.; Lesage, A.; Hediger, S.; Bardet, M.; Meriles, C.A.; Pines, A.; Emsley, L. High-resolution NMR correlation spectra of disordered solids. J. Am. Chem. Soc. 2003, 125, 4376–4380. [Google Scholar] [CrossRef] [PubMed]
- Buléon, A.; Colonna, P.; Planchot, V.; Ball, S. Starch granules: Structure and biosynthesis. Int. J. Biol. Macromol. 1998, 23, 85–112. [Google Scholar] [CrossRef]
- Srichuwong, S.; Sunarti, T.C.; Mishima, T.; Isono, N.; Hisamatsu, M. Starches from different botanical sources I: Contribution of amylopectin fine structure to thermal properties and enzyme digestibility. Carbohydr. Polym. 2005, 60, 529–538. [Google Scholar] [CrossRef]
- Creek, J.A.; Ziegler, G.R.; Runt, J. Amylose crystallization from concentrated aqueous solution. Biomacromolecules 2006, 7, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Glaring, M.A.; Koch, C.B.; Blennow, A. Genotype-specific spatial distribution of starch molecules in the starch granule: A combined CLSM and SEM approach. Biomacromolecules 2006, 7, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Buleon, A.; Duprat, F.; Booy, F.P.; Chanzy, H. Single crystals of amylose with a low degree of polymerization. Carbohydr. Polym. 1984, 4, 161–173. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, J.; Hong, Y.; Liu, G.; Zheng, J.; Gu, Z.; Zhang, P. Impact of amylose content on starch physicochemical properties in transgenic sweet potato. Carbohydr. Polym. 2015, 122, 417–427. [Google Scholar] [CrossRef] [PubMed]
- van Soest, J.J.G.; Essers, P. Influence of amylose-amylopectin ratio on properties of extruded starch plastic sheets. J. Macromol. Sci. Part A Pure Appl. Chem. 1997, 34, 1665–1689. [Google Scholar] [CrossRef]
- Cano, A.; Jiménez, A.; Cháfer, M.; Gónzalez, C.; Chiralt, A. Effect of amylose:amylopectin ratio and rice bran addition on starch films properties. Carbohydr. Polym. 2014, 111, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Klucinec, J.D.; Thompson, D.B. Amylopectin nature and amylose-to-amylopectin ratio as influences on the behavior of gels of dispersed starch. Cereal Chem. 2002, 79, 24–35. [Google Scholar] [CrossRef]
- Wang, S.; Yu, J.; Yu, J. Conformation and location of amorphous and semi-crystalline regions in C-type starch granules revealed by SEM, NMR and XRD. Food Chem. 2008, 110, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Bogracheva, T.Y.; Wang, Y.L.; Hedley, C.L. The effect of water content on the ordered/disordered structures in starches. Biopolymers 2001, 58, 247–259. [Google Scholar] [CrossRef]
- Fontaine, T.; D’Hulst, C.; Maddelein, M.L.; Routier, F.; Pepin, T.M.; Decq, A.; Wieruszeski, J.M.; Delrue, B.; Van den Koornhuyse, N.; Bossu, J.P.; et al. Toward an understanding of the biogenesis of the starch granule. Evidence that Chlamydomonas soluble starch synthase II controls the synthesis of intermediate size glucans of amylopectin. J. Biol. Chem. 1993, 268, 16223–16230. [Google Scholar] [PubMed]
- Delrue, B.; Fontaine, T.; Routier, F.; Decq, A.; Wieruszeski, J.M.; Van Den Koornhuyse, N.; Maddelein, M.L.; Fournet, B.; Ball, S. Waxy Chlamydomonas reinhardtii: Monocellular algal mutants defective in amylose biosynthesis and granule-bound starch synthase activity accumulate a structurally modified amylopectin. J. Bacteriol. 1992, 174, 3612–3620. [Google Scholar] [CrossRef] [PubMed]
- Espada, J. Enzymic synthesis of adenosine diphosphate glucose from glucose 1-phosphate and adenosine triphosphate. J. Biol. Chem. 1962, 237, 3577–3581. [Google Scholar]
- Zabawinski, C.; Van Den Koornhuyse, N.; D’Hulst, C.; Schlichting, R.; Giersch, C.; Delrue, B.; Lacroix, J.M.; Preiss, J.; Ball, S. Starchless mutants of Chlamydomonas reinhardtii lack the small subunit of a heterotetrameric ADP-glucose pyrophosphorylase. J. Bacteriol. 2001, 183, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Surzycki, S. Synchronously grown cultures of Chlamydomonas reinhardi. Methods Enzymol. 1971, 23, 67–73. [Google Scholar] [CrossRef]
- Nocek, J.E.; Tamminga, S. Site of digestion of starch in the gastrointestinal tract of dairy cows and its effect on milk yield and composition. J. Dairy Sci. 1991, 74, 3598–3629. [Google Scholar] [CrossRef]
- Fu, Z.Q.; Wang, L.J.; Li, D.; Zhou, Y.G.; Adhikari, B. The effect of partial gelatinization of corn starch on its retrogradation. Carbohydr. Polym. 2013, 97, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ou, D.; Zheng, L.; Gan, N.; Song, L. Applicability of the fluorescein diacetate assay for metabolic activity measurement of Microcystis aeruginosa (Chroococcales, Cyanobacteria). Phycol. Res. 2011, 59, 200–207. [Google Scholar] [CrossRef]
- Arnold, A.A.; Genard, B.; Zito, F.; Tremblay, R.; Warschawski, D.E.; Marcotte, I. Identification of lipid and saccharide constituents of whole microalgal cells by (1)(3)C solid-state NMR. Biochim. Biophys. Acta 2015, 1848, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Morcombe, C.R.; Zilm, K.W. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003, 162, 479–486. [Google Scholar] [CrossRef]
- Lopez-Rubio, A.; Flanagan, B.M.; Gilbert, E.P.; Gidley, M.J. A novel approach for calculating starch crystallinity and its correlation with double helix content: A combined XRD and NMR study. Biopolymers 2008, 89, 761–768. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C1 | C2 | C3 | C4 | C5 | C6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Amylopectin (A) | 101.8 | → | 73.6 | ||||||||
| 100.6 | → | 72.5 | → | 74.2 | → | 77.9 | → | 71.9 | → | 61.6 | |
| 99.1 | → | 71.9 | |||||||||
| Non reducing end group | 100.5 | → | 72.3 | → | 73.7 | → | 70.4 | → | 73.7 | → | 61.1 |
| Retrograded (B) | 100.9 | → | 72.1 | ↘ | ↗ | 72.4 | → | 61.0 | |||
| 74.1 | → | 77.7 | |||||||||
| 100.1 | → | 72.8 | ↗ | ↘ | 71.5 | → | 61.6 | ||||
| Non reducing end group | 100.1 | → | 72.2 | → | 73.5 | → | 70.4 | → | 73.5 | → | 61.5 |
| Amylose (B) | ↗ | 73.5 | ↘ | ||||||||
| 100.8 | 74.1 | → | 77.8 | → | 71.8 | → | 61.4 | ||||
| ↘ | 72.5 | ↗ | |||||||||
| 99.9 | → | 71.6 | |||||||||
| Non reducing end group | 100.6 | → | 72.6 | → | 73.6 | → | 70.3 | → | 73.6 | → | 61.3 |
| Amorphous | 103.0 | → | 72.7 | → | 75.4 | → | 81.5 | → | 71.5 | → | 60.4 |
| C. reinhardtii wt | 101.5 | → | 72.1 | ||||||||
| Native starch (A) | 100.5 | → | 72.6 | ||||||||
| 99.3 | → | 71.9 | |||||||||
| 75.0 | → | 76.9 | |||||||||
| Non reducing end group | 100.4 | → | 72.4 | → | 73.6 | → | 70.2 | → | 73.6 | → | 61.4 |
| Amorphous | 103.0 | → | 73.0 | → | 75.3 | → | 81.7 | → | 71.5 | → | 60.2 |
| By-product 1 | 92.7 | → | 72.4 | → | 74.9 | → | 76.9 | → | 70.4 | → | 61.4 |
| By-product 2 | 96.7 | → | 74.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poulhazan, A.; Arnold, A.A.; Warschawski, D.E.; Marcotte, I. Unambiguous Ex Situ and in Cell 2D 13C Solid-State NMR Characterization of Starch and Its Constituents. Int. J. Mol. Sci. 2018, 19, 3817. https://doi.org/10.3390/ijms19123817
Poulhazan A, Arnold AA, Warschawski DE, Marcotte I. Unambiguous Ex Situ and in Cell 2D 13C Solid-State NMR Characterization of Starch and Its Constituents. International Journal of Molecular Sciences. 2018; 19(12):3817. https://doi.org/10.3390/ijms19123817
Chicago/Turabian StylePoulhazan, Alexandre, Alexandre A. Arnold, Dror E. Warschawski, and Isabelle Marcotte. 2018. "Unambiguous Ex Situ and in Cell 2D 13C Solid-State NMR Characterization of Starch and Its Constituents" International Journal of Molecular Sciences 19, no. 12: 3817. https://doi.org/10.3390/ijms19123817
APA StylePoulhazan, A., Arnold, A. A., Warschawski, D. E., & Marcotte, I. (2018). Unambiguous Ex Situ and in Cell 2D 13C Solid-State NMR Characterization of Starch and Its Constituents. International Journal of Molecular Sciences, 19(12), 3817. https://doi.org/10.3390/ijms19123817