Intergeneric Relationships within the Early-Diverging Angiosperm Family Nymphaeaceae Based on Chloroplast Phylogenomics

 , ,
, , 
Abstract
1. Introduction
2. Results
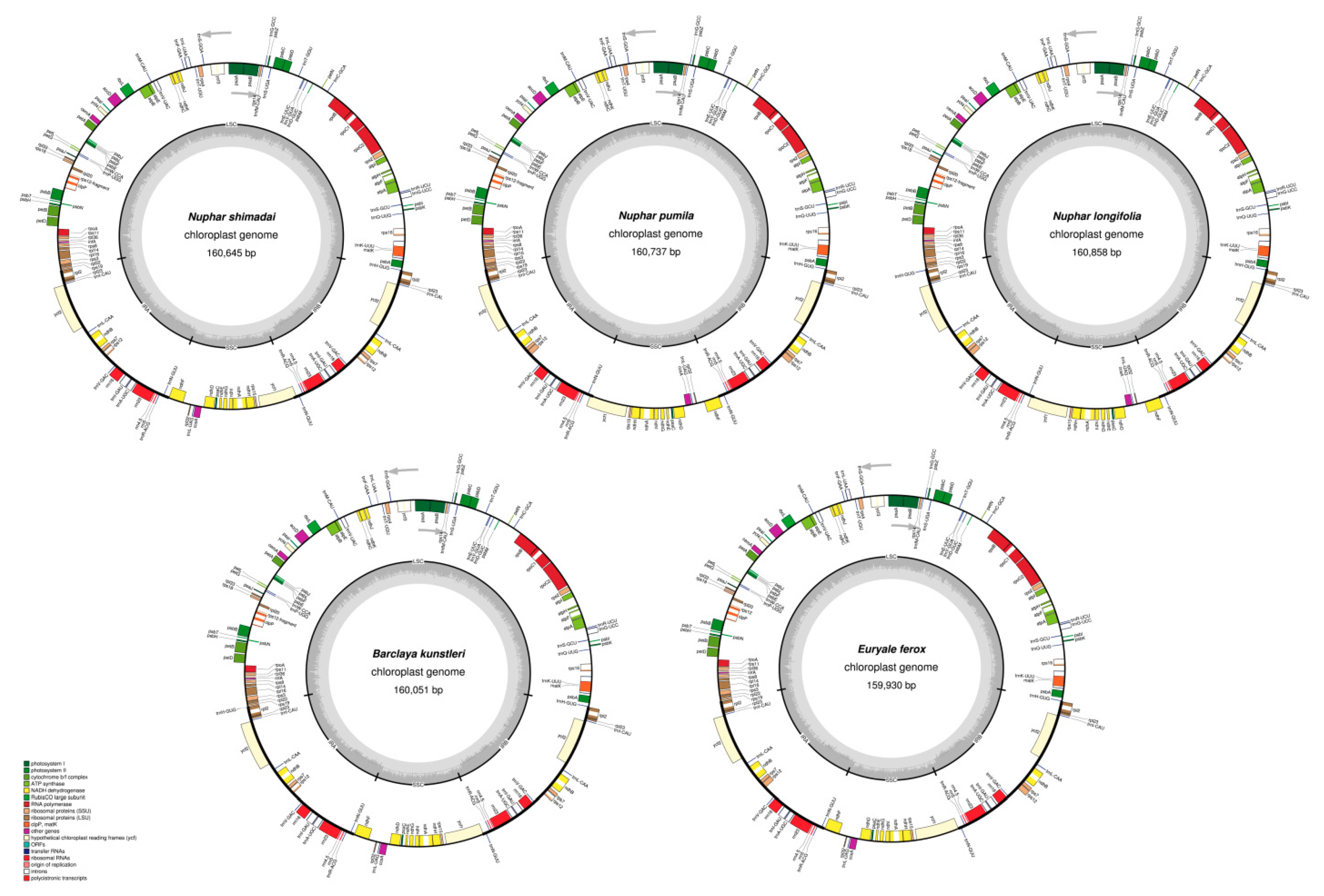
2.1. Structure and Gene Content of the Chloroplast Genomes
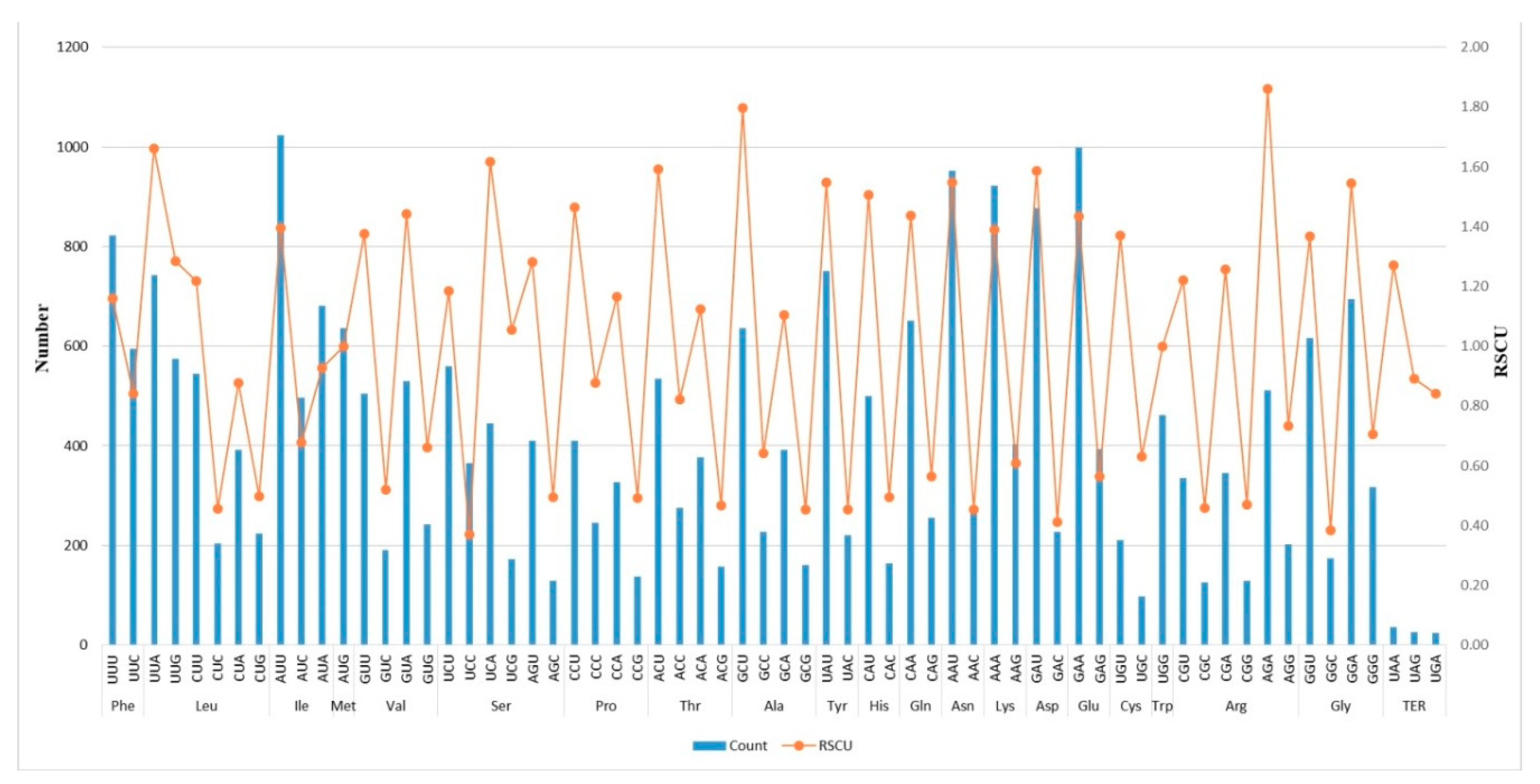
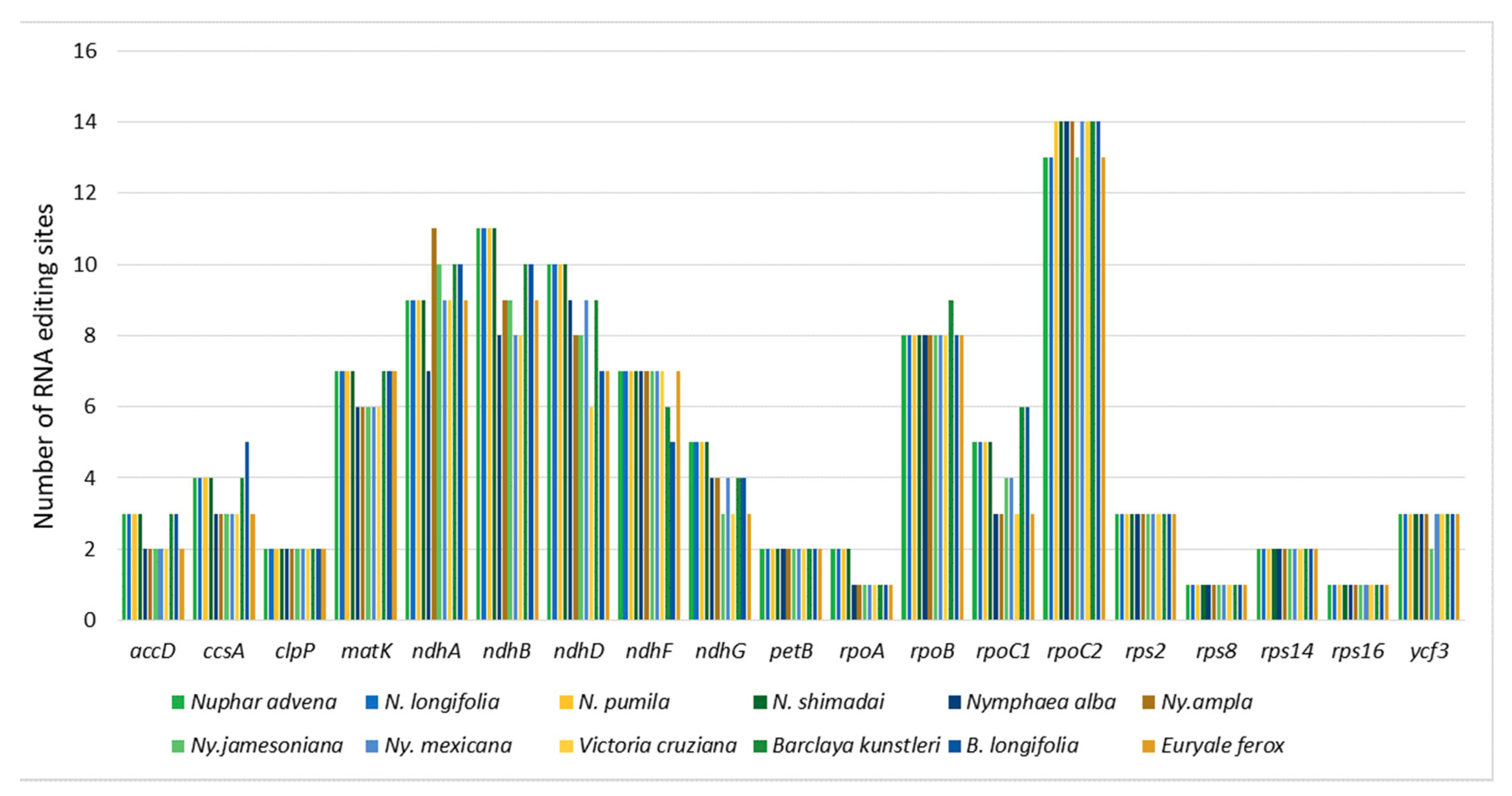
2.2. Codon-Usage, RNA-Editing, and Repetitive-Sequence Analyses
2.3. Inverted Repeats and Genome Comparison
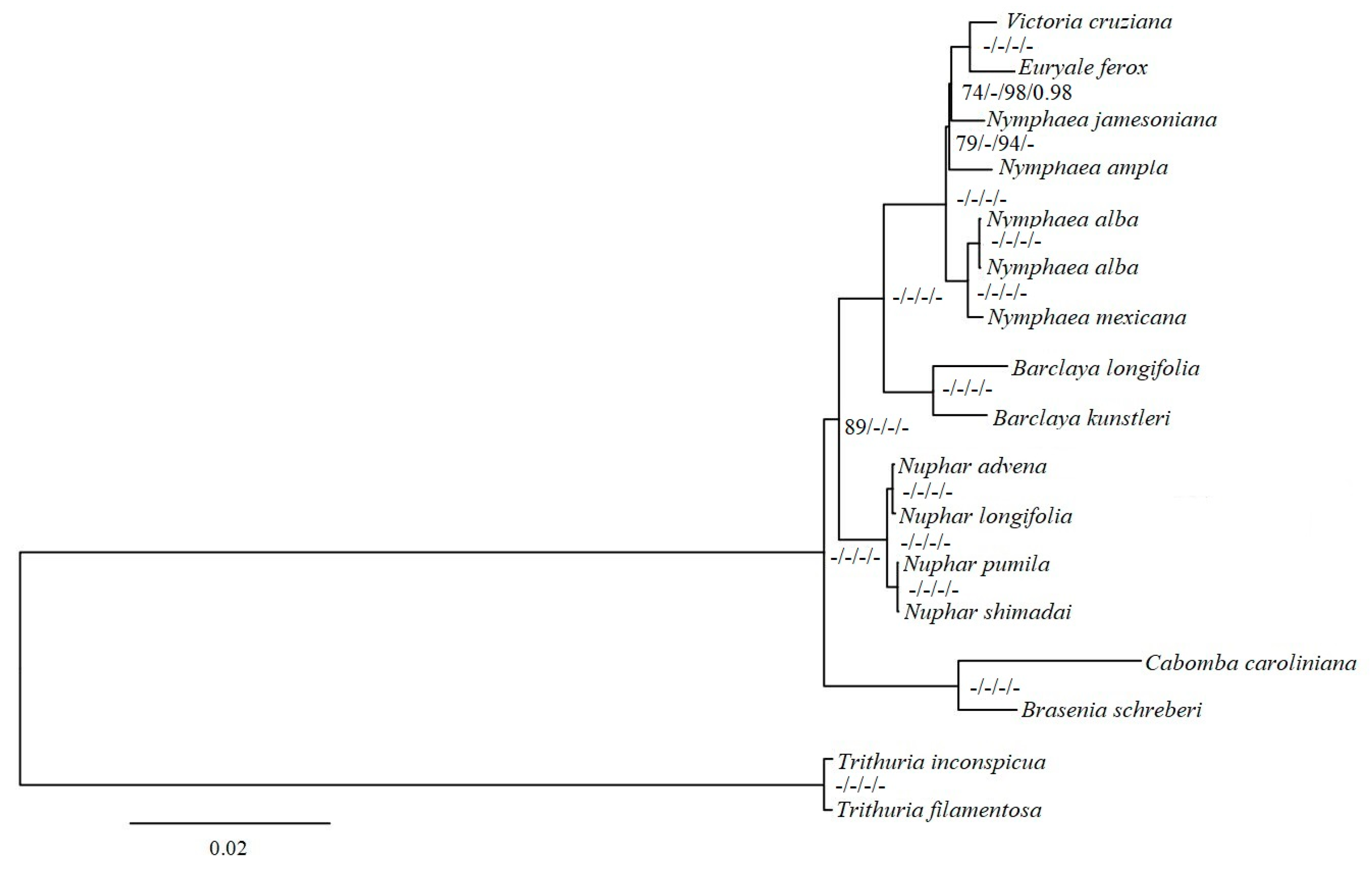
2.4. Phylogenetic Analyses
3. Discussion
3.1. Chloroplast Genome Structure
3.2. Codon-Usage, RNA-Editing, and Repetitive-Sequence Analyses
3.3. Comparative Analyses
3.4. Phylogenetic Inference
4. Materials and Methods
4.1. Plant Material and Genome Sequencing
4.2. Genome Assembly and Annotation
4.3. Chloroplast Genome Comparisons
4.4. Codon-Usage, RNA-Editing and Repetitive Sequences Analyses
4.5. Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IR | Inverted repeat. |
| SSC | Small single copy. |
| LSC | Large single copy. |
| SSR | Simple sequence repeat. |
| PCGS | Protein-coding genes. |
| RNA | Ribonucleic acid |
| RSCU | Relative synonymous codon usage |
| CUB | Codon usage bias |
| UPGMA | Unweighted pair group method with arithmetic mean |
| ML | Maximum likelihoodBI Bayesian inference |
| PP | Posterior probability |
References
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361. [Google Scholar] [CrossRef] [PubMed]
- Gitzendanner, M.A.; Soltis, P.S.; Wong, G.K.S.; Ruhfel, B.R.; Soltis, D.E. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V.; Nikiforova, S.V.; Biggs, P.J.; Zhong, B.; Delange, P.; Martin, W.; Woetzel, S.; Atherton, R.A.; Mclenachan, P.A.; Lockhart, P.J. The evolutionary root of flowering plants. Syst. Biol. 2013, 62, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; Chumley, T.W. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Leebens-Mack, J.; Raubeson, L.A.; Cui, L.; Kuehl, J.V.; Fourcade, M.H.; Chumley, T.W.; Boore, J.L.; Jansen, R.K.; depamphilis, C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the Felsenstein zone. Mol. Biol. Evol. 2005, 22, 1948–1963. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, P.-F.; Li, H.-T.; Yang, J.-B.; Wang, H.; Li, D.-Z. Plastid phylogenomic analyses resolve Tofieldiaceae as the root of the early diverging monocot order Alismatales. Genome Biol. Evol. 2016, 8, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Byng, J.W.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Judd, W.S.; Mabberley, D.J.; Sennikov, A.N.; Soltis, D.E.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Parkinson, C.L.; Adams, K.L.; Palmer, J.D. Multigene analyses identify the three earliest lineages of extant flowering plants. Curr. Biol. 1999, 9, 1485–1491. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E.; Chase, M.W. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 1999, 402, 402. [Google Scholar] [CrossRef] [PubMed]
- Borsch, T.; Hilu, K.W.; Wiersema, J.H.; Löhne, C.; Barthlott, W.; Wilde, V. Phylogeny of Nymphaea (Nymphaeaceae): Evidence from substitutions and microstructural changes in the chloroplast trnT-trnF region. Int. J. Plant Sci. 2007, 168, 639–671. [Google Scholar] [CrossRef]
- Les, D.H.; Garvin, D.K.; Wimpee, C.F. Molecular evolutionary history of ancient aquatic angiosperms. Proc. Natl. Acad. Sci. USA 1991, 88, 10119–10123. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.A.; Qiu, Y.-L.; Endress, P.K.; Friis, E.M. Current perspectives on basal angiosperms: Introduction. Int. J. Plant Sci. 2000, 161, S1–S2. [Google Scholar] [CrossRef]
- Friedman, W.E. Hydatellaceae are water lilies with gymnospermous tendencies. Nature 2008, 453, 94. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.M.; Rai, H.S.; Doyle, J.A.; Endress, P.K.; Mathews, S.; Marchant, A.D.; Briggs, B.G.; Graham, S.W. Hydatellaceae identified as a new branch near the base of the angiosperm phylogenetic tree. Nature 2007, 446, 312. [Google Scholar] [CrossRef] [PubMed]
- Biswal, D.K.; Debnath, M.; Kumar, S.; Tandon, P. Phylogenetic reconstruction in the Order Nymphaeales: ITS2 secondary structure analysis and in silico testing of Maturase K (matK) as a potential marker for DNA bar coding. Proc. BMC Bioinform. 2012, 13, S26. [Google Scholar]
- Gruenstaeudl, M.; Nauheimer, L.; Borsch, T. Plastid genome structure and phylogenomics of Nymphaeales: Conserved gene order and new insights into relationships. Plant Syst. Evol. 2017, 303, 1251–1270. [Google Scholar] [CrossRef]
- Loehne, C.; Borsch, T.; Wiersema, J.H. Phylogenetic analysis of Nymphaeales using fast-evolving and noncoding chloroplast markers. Bot. J. Linn. Soc. 2007, 154, 141–163. [Google Scholar] [CrossRef]
- Nandi, O.I.; Chase, M.W.; Endress, P.K. A combined cladistic analysis of angiosperms using rbcL and non-molecular data sets. Ann. Mo. Bot. Gard. 1998, 137–214. [Google Scholar] [CrossRef]
- Sokoloff, D.D.; Remizowa, M.V.; Macfarlane, T.D.; Rudall, P.J. Classification of the early-divergent angiosperm family Hydatellaceae: One genus instead of two, four new species and sexual dimorphism in dioecious taxa. Taxon 2008, 57, 179–200. [Google Scholar]
- Christenhusz, M.J.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Gruenstaeudl, M.; Gerschler, N.; Borsch, T. Bioinformatic workflows for generating complete Plastid genome sequences—An example from Cabomba (Cabombaceae) in the context of the phylogenomic analysis of the water-lily clade. Life 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Moore, M.J.; Zhang, S.; Soltis, P.S.; Soltis, D.E.; Zhao, T.; Meng, A.; Li, X.; Li, J.; Wang, H. Phylogenomic and structural analyses of 18 complete plastomes across nearly all families of early-diverging eudicots, including an angiosperm-wide analysis of IR gene content evolution. Mol. Phylogenet. Evol. 2016, 96, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Ming, Z.; Junrui, W.; Li, F.; Sha, L.; Hongbo, P.; Lan, Q.; Jing, L.; Yan, S.; Weihua, Q.; Lifang, Z. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.W.; Olmstead, R.G.; Barrett, S.C. Rooting phylogenetic trees with distant outgroups: A case study from the commelinoid monocots. Mol. Biol. Evol. 2002, 19, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; dePamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J. Section I-Comparing macromolecules: Exploring biological diversity-subsection C-the genomes-20 methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–383. [Google Scholar]
- Palmer, J.D. Plastid chromosomes: Structure and evolution. Mol. Biol. Plastids 1991, 7, 5–53. [Google Scholar]
- Goremykin, V.V.; Hirsch-Ernst, K.I.; Wölfl, S.; Hellwig, F.H. Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol. Biol. Evol. 2003, 20, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium× hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wu, J.; Yang, H.; Bao, Q. Codon usage patterns in Corynebacterium glutamicum: Mutational bias, natural selection and amino acid conservation. Comp. Funct. Genom. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Uddin, A. Indices of Codon Usage Bias. Proteom. Bioinform. 2017, 10, 6. [Google Scholar] [CrossRef]
- Liu, Q.; Xue, Q. Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 2005, 84, 55–62. [Google Scholar] [CrossRef]
- Zhou, M.; Long, W.; Li, X. Patterns of synonymous codon usage bias in chloroplast genomes of seed plants. For. Stud. China 2008, 10, 235. [Google Scholar] [CrossRef]
- Schmitz-Linneweber, C.; Barkan, A. RNA splicing and RNA editing in chloroplasts. In Cell and Molecular Biology of Plastids; Springer: Berlin/Heidelber, Germany, 2007; pp. 213–248. [Google Scholar]
- Sugiura, M. RNA editing in chloroplasts. In RNA Editing; Springer: Berlin/Heidelber, Germany, 2008; pp. 123–142. [Google Scholar]
- Freyer, R.; Kiefer-Meyer, M.-C.; Kössel, H. Occurrence of plastid RNA editing in all major lineages of land plants. Proc. Natl. Acad. Sci. USA 1997, 94, 6285–6290. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.G.; Awasthi, P.; Bedi, Y.S. Analysis of SSR dynamics in chloroplast genomes of Brassicaceae family. Bioinformation 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Lozano, J.J.; Ortiz, A.R. Comparative analysis of chloroplast genomes: Functional annotation, genome-based phylogeny, and deduced evolutionary patterns. Genome Res. 2002, 12, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, G.M.; Downie, S.R. Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst. Bot. 2000, 648–667. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Jian, S.; Soltis, P.S.; Gitzendanner, M.A.; Moore, M.J.; Li, R.; Hendry, T.A.; Qiu, Y.-L.; Dhingra, A.; Bell, C.D.; Soltis, D.E. Resolving an ancient, rapid radiation in Saxifragales. Syst. Biol. 2008, 57, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Borsch, T.; Löhne, C.; Wiersema, J. Phylogeny and evolutionary patterns in Nymphaeales: Integrating genes, genomes and morphology. Taxon 2008, 57, 1052. [Google Scholar]
- Wheeler, W.C. Nucleic acid sequence phylogeny and random outgroups. Cladistics 1990, 6, 363–367. [Google Scholar] [CrossRef]
- Goremykin, V.; Hirsch-Ernst, K.; Wölfl, S.; Hellwig, F. The chloroplast genome of the “basal” angiosperm Calycanthus fertilis–structural and phylogenetic analyses. Plant Syst. Evol. 2003, 242, 119–135. [Google Scholar] [CrossRef]
- Stefanović, S.; Rice, D.W.; Palmer, J.D. Long branch attraction, taxon sampling, and the earliest angiosperms: Amborella or monocots? BMC Evol. Biol. 2004, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Les, D.H.; Schneider, E.L.; Padgett, D.J.; Soltis, P.S.; Soltis, D.E.; Zanis, M. Phylogeny, classification and floral evolution of water lilies (Nymphaeaceae; Nymphaeales): A synthesis of non-molecular, rbcL, matK, and 18S rDNA data. Syst. Bot. 1999, 28–46. [Google Scholar] [CrossRef]
- Padgett, D.J. A monograph of Nuphar (Nymphaeaceae). Rhodora 2007, 109, 1–95. [Google Scholar] [CrossRef]
- Padgett, D.J.; Les, D.H.; Crow, G.E. Phylogenetic relationships in Nuphar (Nymphaeaceae): Evidence from morphology, chloroplast DNA, and nuclear ribosomal DNA. Am. J. Bot. 1999, 86, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Williamson, P.S.; Schneider, E.L. Floral aspects of Barclaya (Nymphaeaceae): Pollination, ontogeny and structure. In Early Evolution of Flowers; Springer: Vienna, Austria, 1994; pp. 159–173. [Google Scholar]
- Schneider, E.; Williamson, P. Nymphaeaceae. In Flowering Plants · Dicotyledons: Magnoliid, Hamamelid and Caryophyllid Families; Kubitzki, K., Rohwer, J.G., Bittrich, V., Eds.; Springer: Berlin/Heidelber, Germany, 1993; pp. 486–493. [Google Scholar]
- Borsch, T.; Wiersema, J.H.; Hellquist, C.B.; Löhne, C.; Govers, K. Speciation in North American water lilies: Evidence for the hybrid origin of the newly discovered Canadian endemic Nymphaea loriana sp. nov.(Nymphaeaceae) in a past contact zone. Botany 2014, 92, 867–882. [Google Scholar] [CrossRef]
- Thorne, R.F. Classification and geography of the flowering plants. Bot. Rev. 1992, 58, 225–327. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Organism | Barclaya kunstleri | Euryale ferox Salisb. | Nuphar longifolia (Michx.) Sm. | Nuphar pumila (Timm) DC. | Nuphar shimadai Hayata |
|---|---|---|---|---|---|
| GenBank accession number | KY392762 | KY392765 | MH050795 | MH050796 | MH050797 |
| Genome size (bp) | 160,051 | 159,930 | 160,858 | 160,737 | 160,645 |
| Large single copy (LSC) length (bp) | 90,026 | 89,677 | 90,375 | 90,610 | 90,551 |
| Small single copy (SSC) length (bp) | 19,325 | 20,201 | 18,811 | 18,865 | 18,830 |
| Inverted repeat (IR) length (bp) | 25,350 | 25,026 | 25,836 | 25,631 | 25,632 |
| Number of genes | 113 | 113 | 113 | 113 | 113 |
| Number of protein-coding genes (duplicated in IR) | 79 (6) | 79 (6) | 79 (6) | 79 (6) | 79 (6) |
| Number of tRNA genes (duplicated in IR) | 30 (7) | 30 (7) | 30 (8) | 30 (7) | 30 (7) |
| Number of rRNA genes (duplicated in IR) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) |
| Number of genes with one intron (two introns) | 15 (3) | 15 (3) | 15 (3) | 15 (3) | 15 (3) |
| Proportion of coding to noncoding regions | 0.68 | 0.68 | 0.69 | 0.68 | 0.69 |
| Average gene density (genes/kb) | 0.82 | 0.82 | 0.83 | 0.82 | 0.82 |
| GC content (%) | 39.1 | 39.1 | 39.1 | 39.1 | 39.1 |
| Category | Gene Type | Gene | ||||||
|---|---|---|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA | rrn16 | rrn23 | rrn4.5 | rrn5 | |||
| Transfer RNA | trnA-UGC * | trnfM-CAU | trnI-GAU * | trnM-CAU | trnR-ACG | trnS-UGA | ||
| trnC-GCA | trnG-GCC | trnK-UUU * | trnN-GUU | trnW-CCA | trnT-GGU | |||
| trnD-GUC | trnG-UCC * | trnL-CAA | trnY-GUA | trnR-UCU | trnT-UGU | |||
| trnE-UUC | trnH-GUG | trnL-UAA * | trnP-UGG | trnS-GCU | trnV-GAC | |||
| trnF-GAA | trnI-CAU | trnL-UAG | trnQ-UUG | trnS-GGA | trnV-UAC * | |||
| Small ribosomal units | rps11 | rps12 | rps14 | rps15 | rps16 * | rps18 | ||
| rps19 | rps2 | rps3 | rps4 | rps7 | rps8 | |||
| Large ribosomal units | rpl14 | rpl16 | rpl2 * | rpl20 | rpl22 | rpl23 | rpl32 | |
| rpl33 | rpl36 | |||||||
| RNA polymerase subunits | rpoA | rpoB | rpoC1 * | rpoC2 | ||||
| translation initiation factor | infA | |||||||
| Photosynthesis genes | NADH dehydrogenase | ndhA * | NdhB * | ndhC | ndhD | ndhE | ndhF | |
| ndhG | ndhH | ndhI | ndhJ | ndhK | ||||
| photosystem I | psaA | psaB | psaC | psaI | psaJ | ycf3 ** | ycf4 | |
| photosystem II | psbA | psbB | psbC | psbD | psbE | psbF | psbH | |
| psbI | psbJ | psbK | psbL | psbM | psbN | psbT | ||
| psbZ | ||||||||
| cytochrome b/f complex | petA | petB | petD | petG | petL | petN | ||
| ATP synthase | atpA | atpB | atpE | atpF * | atpH | atpI | ||
| Large subunit of rubisco | rbcL | |||||||
| Other genes | Maturase | matK | ||||||
| Protease | clpP ** | |||||||
| Acetyl-CoA-carboxylase sub-unit | accD | |||||||
| Envelope membrane protein | cemA | |||||||
| Component of TIC complex | ycf1 | |||||||
| c-type cytochrome synthesis | ccsA | |||||||
| Unknown | hypothetical genes reading frames | ycf2 | ||||||
| Nuphar advena | Nuphar longifolia | Nuphar pumila | Nuphar shimadai | Nymphaea alba | Nymphaea ampla | Nymphaea jamesoniana | Nymphaea mexicana | Victoria cruziana | Euryale ferox | Barclaya kunstleri | Barclaya longifolia |
|---|---|---|---|---|---|---|---|---|---|---|---|
| accD3 | accD3 | accD3 | accD3 | accD2 | accD2 | accD2 | accD2 | accD2 | accD2 | accD3 | accD3 |
| atpA | atpA | atpA | atpA | AtpA | atpA | atpA | atpA | atpA | atpA | atpA | |
| atpB | atpB | atpB | atpB | atpB | atpB | atpB | atpB | ||||
| atpF | |||||||||||
| atpI | atpI | atpI | atpI | atpI | atpI | atpI | atpI | ||||
| ccsA4 | ccsA4 | ccsA4 | ccsA4 | ccsA3 | ccsA3 | ccsA3 | ccsA3 | ccsA3 | ccsA3 | ccsA4 | ccsA5 |
| clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 | clpP2 |
| matK7 | matK7 | matK7 | matK7 | matK6 | matK6 | matK6 | matK6 | matK6 | matK7 | matK7 | matK7 |
| ndhA9 | ndhA9 | ndhA9 | ndhA9 | ndhA7 | ndhA11 | ndhA10 | ndhA9 | ndhA9 | ndhA9 | ndhA10 | ndhA10 |
| ndhB11 | ndhB11 | ndhB11 | ndhB11 | ndhB8 | ndhB9 | ndhB9 | ndhB8 | ndhB8 | ndhB9 | ndhB10 | ndhB10 |
| ndhD10 | ndhD10 | ndhD10 | ndhD10 | ndhD9 | ndhD8 | ndhD8 | ndhD9 | ndhD6 | ndhD7 | ndhD9 | ndhD7 |
| ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF7 | ndhF6 | ndhF5 |
| ndhG5 | ndhG5 | ndhG5 | ndhG5 | ndhG4 | ndhG4 | ndhG3 | ndhG4 | ndhG3 | ndhG3 | ndhG4 | ndhG4 |
| petB2 | petB2 | petB2 | petB2 | petB2 | petB2 | petB2 | petB2 | petB2 | petB2 | petB2 | petB2 |
| petD | |||||||||||
| petG | petG | petG | petG | petG | petG | ||||||
| psbE | psbE | psbE | psbE | psbE | psbE | psbE | psbE | psbE | psbE | psbE | |
| psbF | psbF | psbF | psbF | psbF | psbF | ||||||
| psbL | psbL | psbL | psbL | psbL | psbL | psbL | psbL | psbL | psbL | ||
| rpl2 | rpl2 | rpl2 | rpl2 | rpl2 | rpl2 | rpl2 | rpl2 | ||||
| rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | rpl20 | |
| rpoA2 | rpoA2 | rpoA2 | rpoA2 | rpoA1 | rpoA1 | rpoA1 | rpoA1 | rpoA1 | rpoA1 | rpoA1 | rpoA1 |
| rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 | rpoB8 |
| rpoC15 | rpoC15 | rpoC15 | rpoC15 | rpoC13 | rpoC13 | rpoC14 | rpoC14 | rpoC13 | rpoC13 | rpoC16 | rpoC16 |
| rpoC213 | rpoC213 | rpoC214 | rpoC214 | rpoC214 | rpoC214 | rpoC213 | rpoC214 | rpoC214 | rpoC213 | rpoC214 | rpoC214 |
| rps23 | rps23 | rps23 | rps23 | rps23 | rps23 | rps23 | rps23 | rps23 | rps23 | rps23 | rps23 |
| rps81 | rps81 | rps81 | rps81 | rps81 | rps81 | rps81 | rps81 | rps81 | rps81 | rps81 | rps81 |
| rps142 | rps142 | rps142 | rps142 | rps142 | rps142 | rps142 | rps142 | rps142 | rps142 | rps142 | rps142 |
| rps161 | rps161 | rps161 | rps161 | rps161 | rps161 | rps161 | rps161 | rps161 | rps161 | rps161 | rps161 |
| ycf33 | ycf33 | ycf33 | ycf33 | ycf33 | ycf33 | ycf32 | ycf33 | ycf33 | ycf33 | ycf33 | ycf33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, D.; Gichira, A.W.; Li, Z.; Nzei, J.M.; Guo, Y.; Wang, Q.; Chen, J. Intergeneric Relationships within the Early-Diverging Angiosperm Family Nymphaeaceae Based on Chloroplast Phylogenomics. Int. J. Mol. Sci. 2018, 19, 3780. https://doi.org/10.3390/ijms19123780
He D, Gichira AW, Li Z, Nzei JM, Guo Y, Wang Q, Chen J. Intergeneric Relationships within the Early-Diverging Angiosperm Family Nymphaeaceae Based on Chloroplast Phylogenomics. International Journal of Molecular Sciences. 2018; 19(12):3780. https://doi.org/10.3390/ijms19123780
Chicago/Turabian StyleHe, Dingxuan, Andrew W. Gichira, Zhizhong Li, John M. Nzei, Youhao Guo, Qingfeng Wang, and Jinming Chen. 2018. "Intergeneric Relationships within the Early-Diverging Angiosperm Family Nymphaeaceae Based on Chloroplast Phylogenomics" International Journal of Molecular Sciences 19, no. 12: 3780. https://doi.org/10.3390/ijms19123780
APA StyleHe, D., Gichira, A. W., Li, Z., Nzei, J. M., Guo, Y., Wang, Q., & Chen, J. (2018). Intergeneric Relationships within the Early-Diverging Angiosperm Family Nymphaeaceae Based on Chloroplast Phylogenomics. International Journal of Molecular Sciences, 19(12), 3780. https://doi.org/10.3390/ijms19123780