Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis


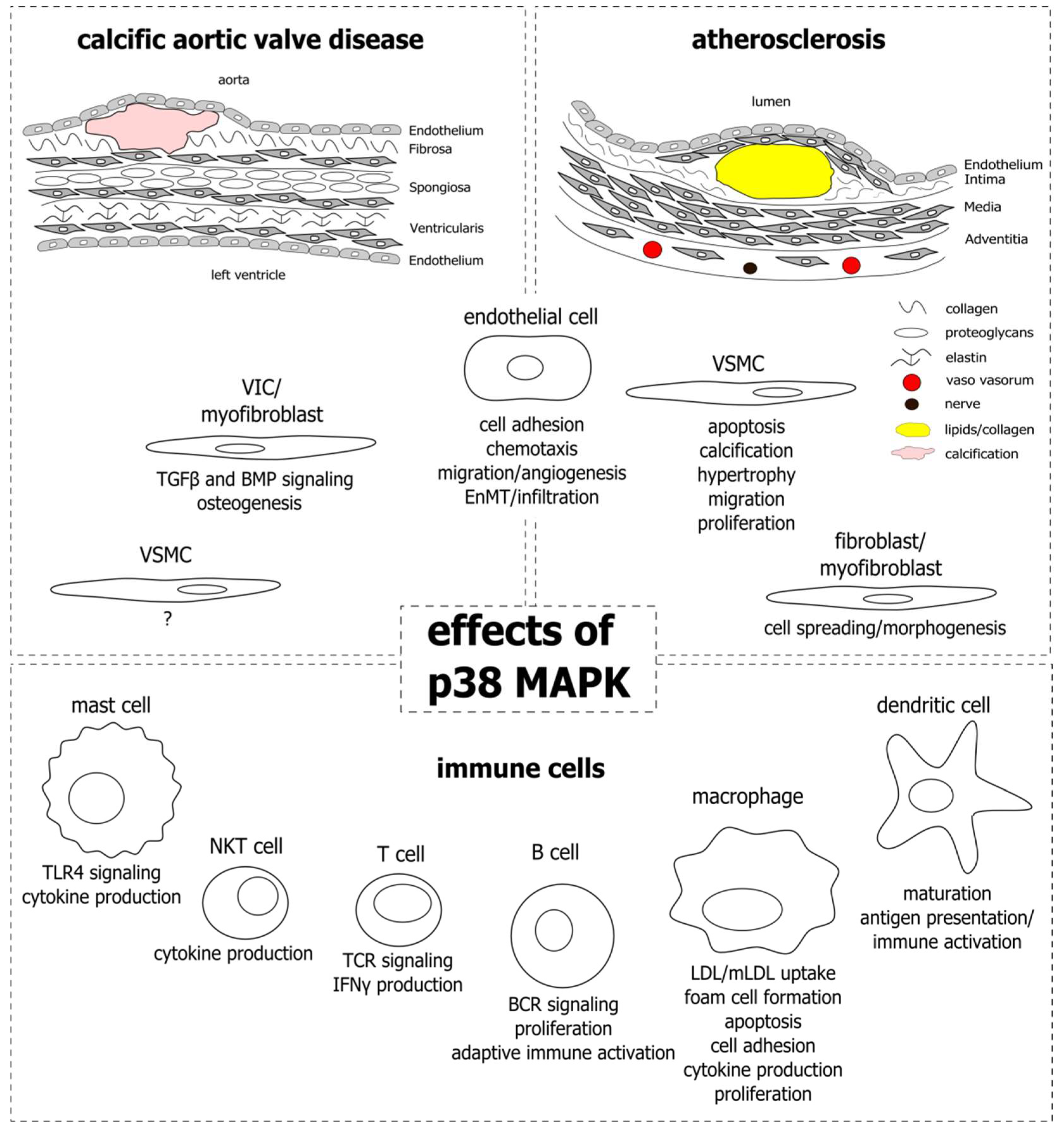{kind=link}
Abstract
1. Introduction
2. p38 MAPK Signaling
3. Endothelial Cells
4. Smooth Muscle Cells
5. Aortic Valve Interstitial Cells, Myofibroblasts, and Vascular Fibroblasts
6. Monocytes and Macrophages
7. Other Immune Cells: Mast Cells, T Cells, Natural Killer T Cells, B Cells, and Dendritic Cells
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ang II | angiotensin II |
| ApoE | apolipoprotein E |
| AVS | aortic valve stenosis |
| BMP2 | bone morphogenic protein 2 |
| CAVD | calcific aortic valve disease |
| DC | dendritic cell |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| eLDL | enzymatically modified LDL |
| EnMT | endothelial-to-mesenchymal transition |
| ER | endoplasmic reticulum |
| ERK | extracellular signal regulated kinase |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GWAS | genome-wide association study |
| HDL | high-density lipoprotein |
| HUVEC | human umbilical-vein endothelial cell |
| ICAM-1 | intercellular adhesion molecule 1 |
| IL | interleukin |
| JNK | c-Jun N-terminal kinase |
| IFN | interferon |
| LDL | low-density lipoprotein |
| Lp(a) | lipoprotein a |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MAPKAPK2 | MAPK-activated protein kinase 2 |
| MAPKK | MAPK kinase |
| MAPKKK | MAPK kinase kinase |
| MCP-1 | monocyte-chemoattractant protein 1 |
| MFB | myofibroblast |
| mLDL | modified LDL |
| MMP | matrix metalloproteinase |
| NKT | natural killer T cell |
| NO | nitric oxide |
| oxHDL | oxidized HDL |
| oxLDL | oxidized LDL |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| SMAD | small mothers against decapentaplegic |
| SMC | smooth muscle cell |
| TCR | T cell receptor |
| TGFβ | transforming growth factor beta |
| TLR4 | toll-like receptor 4 |
| VCAM-1 | vascular cell adhesion protein 1 |
| VIC | valve interstitial cell |
| VSMC | vascular smooth muscle cell |
References
- World Health Organization. Cardiovascular Diseases. Available online: http://www.euro.who.int/en/health-topics/noncommunicable-diseases/cardiovascular-diseases (accessed on 10 September 2018).
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Serban, C.; Sahebkar, A.; Mikhailidis, D.P.; Ursoniu, S.; Ray, K.K.; Rysz, J.; Toth, P.P.; Muntner, P.; Mosteoru, S.; et al. Impact of statin therapy on coronary plaque composition: A systematic review and meta-analysis of virtual histology intravascular ultrasound studies. BMC Med. 2015, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sasaki, T.; Cheng, X.W.; Iguchi, A.; Sato, K.; Kuzuya, M. Statin prevents plaque disruption in apoE-knockout mouse model through pleiotropic effect on acute inflammation. Atherosclerosis 2009, 206, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Osnabrugge, R.L.J.; Mylotte, D.; Head, S.J.; van Mieghem, N.M.; Nkomo, V.T.; LeReun, C.M.; Bogers, A.J.J.C.; Piazza, N.; Kappetein, A.P. Aortic stenosis in the elderly: Disease prevalence and number of candidates for transcatheter aortic valve replacement: A meta-analysis and modeling study. J. Am. Coll. Cardiol. 2013, 62, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A.; Bäck, M. Alcohol consumption, cigarette smoking and incidence of aortic valve stenosis. J. Intern. Med. 2017, 282, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A.; Håkansson, N.; Bäck, M. Overall and abdominal obesity and incident aortic valve stenosis: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, K.; Mohseni, H.; Kiran, A.; Tran, J.; Nazarzadeh, M.; Rahimian, F.; Woodward, M.; Dwyer, T.; MacMahon, S.; Otto, C.M. Elevated blood pressure and risk of aortic valve disease: A cohort analysis of 5.4 million UK adults. Eur. Heart J. 2018, 39, 3596–3603. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef]
- Guerraty, M.A.; Chai, B.; Hsu, J.Y.; Ojo, A.O.; Gao, Y.; Yang, W.; Keane, M.G.; Budoff, M.J.; Mohler, E.R. Relation of aortic valve calcium to chronic kidney disease (from the Chronic Renal Insufficiency Cohort Study). Am. J. Cardiol. 2015, 115, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.; Wong, N.D.; Kronmal, R.; Takasu, J.; Shavelle, D.M.; Probstfield, J.L.; Bertoni, A.G.; Budoff, M.J.; O’Brien, K.D. Features of the metabolic syndrome and diabetes mellitus as predictors of aortic valve calcification in the Multi-Ethnic Study of Atherosclerosis. Circulation 2006, 113, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Zhu, Q.M.; Emdin, C.A.; Chaffin, M.; Horner, S.; McMillan, B.J.; Leed, A.; Weale, M.E.; Spencer, C.C.A.; Aguet, F.; et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat. Genet. 2017, 49, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Howson, J.M.M.; Zhao, W.; Barnes, D.R.; Ho, W.-K.; Young, R.; Paul, D.S.; Waite, L.L.; Freitag, D.F.; Fauman, E.B.; Salfati, E.L.; et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 2017, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Thorleifsson, G.; Gretarsdottir, S.; Stefansson, O.A.; Tragante, V.; Thorolfsdottir, R.B.; Jonsdottir, I.; Bjornsson, T.; Steinthorsdottir, V.; Verweij, N.; et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat. Commun. 2018, 9, 987. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; Franke, U.F.W.; et al. Lipoprotein(a) Associated Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Bozbas, H.; Yildirir, A.; Atar, I.; Pirat, B.; Eroglu, S.; Aydinalp, A.; Ozin, B.; Muderrisoglu, H. Effects of serum levels of novel atherosclerotic risk factors on aortic valve calcification. J. Heart Valve Dis. 2007, 16, 387–393. [Google Scholar] [PubMed]
- Gotoh, T.; Kuroda, T.; Yamasawa, M.; Nishinaga, M.; Mitsuhashi, T.; Seino, Y.; Nagoh, N.; Kayaba, K.; Yamada, S.; Matsuo, H. Correlation between lipoprotein(a) and aortic valve sclerosis assessed by echocardiography (the JMS Cardiac Echo and Cohort Study). Am. J. Cardiol. 1995, 76, 928–932. [Google Scholar] [CrossRef]
- Rajamannan, N.M. Calcific aortic stenosis: Lessons learned from experimental and clinical studies. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation and Atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.V.; Otto, C.M. Spectrum of Calcific Aortic Valve Disease: Pathogenesis, Disease Progression, and Treatment Strategies. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110 Pt 3, 357–368. [Google Scholar]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Bonney, E.A. Mapping out p38MAPK. Am. J. Reprod. Immunol. 2017, 77. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M. Enzymatically modified LDL, atherosclerosis and beyond: Paving the way to acceptance. Front. Biosci. 2018, 23, 1257–1271. [Google Scholar] [CrossRef]
- Zhu, Y.; Liao, H.; Wang, N.; Ma, K.S.; Verna, L.K.; Shyy, J.Y.; Chien, S.; Stemerman, M.B. LDL-activated p38 in endothelial cells is mediated by Ras. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Pietersma, A.; Tilly, B.C.; Gaestel, M.; de Jong, N.; Lee, J.C.; Koster, J.F.; Sluiter, W. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem. Biophys. Res. Commun. 1997, 230, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.; Kilian, K.; Gillitzer, R.; Kunz, M.; Yoshimura, T.; Bröcker, E.B.; Rapp, U.R.; Ludwig, S. The MKK6/p38 stress kinase cascade is critical for tumor necrosis factor-alpha-induced expression of monocyte-chemoattractant protein-1 in endothelial cells. Blood 1999, 93, 857–865. [Google Scholar] [PubMed]
- Liu, L.; Craig, A.W.; Meldrum, H.D.; Marcovina, S.M.; Elliott, B.E.; Koschinsky, M.L. Apolipoprotein(a) stimulates vascular endothelial cell growth and migration and signals through integrin alphaVbeta3. Biochem. J. 2009, 418, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- McMullen, M.E.; Bryant, P.W.; Glembotski, C.C.; Vincent, P.A.; Pumiglia, K.M. Activation of p38 has opposing effects on the proliferation and migration of endothelial cells. J. Biol. Chem. 2005, 280, 20995–21003. [Google Scholar] [CrossRef] [PubMed]
- Mohler, E.R.; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone formation and inflammation in cardiac valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Lerman, L.O.; Mukhopadhyay, D.; Napoli, C.; Lerman, A. Angiogenesis in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Borbiev, T.; Birukova, A.; Liu, F.; Nurmukhambetova, S.; Gerthoffer, W.T.; Garcia, J.G.N.; Verin, A.D. p38 MAP kinase-dependent regulation of endothelial cell permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L911–L918. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Gomaraschi, M.; Basilico, N.; Sisto, F.; Taramelli, D.; Eligini, S.; Colli, S.; Sirtori, C.R.; Franceschini, G.; Calabresi, L. High-density lipoproteins attenuate interleukin-6 production in endothelial cells exposed to pro-inflammatory stimuli. Biochim. Biophys. Acta 2005, 1736, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, J.; Escudero, I.; Badimon, L. Simvastatin potenciates PGI(2) release induced by HDL in human VSMC: Effect on Cox-2 up-regulation and MAPK signalling pathways activated by HDL. Atherosclerosis 2004, 174, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Jin, W.; Chen, H. oxHDL decreases the expression of CD36 on human macrophages through PPARgamma and p38 MAP kinase dependent mechanisms. Mol. Cell. Biochem. 2010, 342, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Banfi, C.; Pirillo, A.; Tremoli, E.; Hamsten, A.; Catapano, A.L.; Eriksson, P. Oxidised-HDL3 induces the expression of PAI-1 in human endothelial cells. Role of p38MAPK activation and mRNA stabilization. Br. J. Haematol. 2004, 127, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Callegari, E.; Inoue, H.; Catapano, A.L. HDL3 induces cyclooxygenase-2 expression and prostacyclin release in human endothelial cells via a p38 MAPK/CRE-dependent pathway: Effects on COX-2/PGI-synthase coupling. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Wylie-Sears, J.; Aikawa, E.; Levine, R.A.; Yang, J.-H.; Bischoff, J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, K.; Alford, P.W.; Wylie-Sears, J.; Goss, J.A.; Grosberg, A.; Bischoff, J.; Aikawa, E.; Levine, R.A.; Parker, K.K. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc. Natl. Acad. Sci. USA 2011, 108, 19943–19948. [Google Scholar] [CrossRef] [PubMed]
- Sumpio, B.E.; Yun, S.; Cordova, A.C.; Haga, M.; Zhang, J.; Koh, Y.; Madri, J.A. MAPKs (ERK1/2, p38) and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J. Biol. Chem. 2005, 280, 11185–11191. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Duzgun, S.A.; Ikeda, M.; Kito, H.; Akasaka, N.; Sasajima, T.; Sumpio, B.E. Endothelial cell response to different mechanical forces. J. Vasc. Surg. 2000, 32, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Akasaka, N.; Kito, H.; Ikeda, M.; Gahtan, V.; Sasajima, T.; Sumpio, B.E. Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H189–H197. [Google Scholar] [CrossRef] [PubMed]
- Kardakaris, R.; Gareus, R.; Xanthoulea, S.; Pasparakis, M. Endothelial and macrophage-specific deficiency of P38α MAPK does not affect the pathogenesis of atherosclerosis in ApoE−/− mice. PLoS ONE 2011, 6, e21055. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2017, 25, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Richardson, P.D.; Woolf, N.; Katz, D.R.; Mann, J. Risk of thrombosis in human atherosclerotic plaques: Role of extracellular lipid, macrophage, and smooth muscle cell content. Br. Heart J. 1993, 69, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.M. Apoptosis in the atherosclerotic plaque: Quantitative and qualitative aspects. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Nishio, E.; Arimura, S.; Watanabe, Y. Oxidized LDL induces apoptosis in cultured smooth muscle cells: A possible role for 7-ketocholesterol. Biochem. Biophys. Res. Commun. 1996, 223, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Chau, L. Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2001, 280, C709–C718. [Google Scholar] [CrossRef] [PubMed]
- Jovinge, S.; Crisby, M.; Thyberg, J.; Nilsson, J. DNA fragmentation and ultrastructural changes of degenerating cells in atherosclerotic lesions and smooth muscle cells exposed to oxidized LDL in vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Jing, Q.; Xin, S.M.; Cheng, Z.J.; Zhang, W.B.; Zhang, R.; Qin, Y.W.; Pei, G. Activation of p38 mitogen-activated protein kinase by oxidized LDL in vascular smooth muscle cells: Mediation via pertussis toxin-sensitive G proteins and association with oxidized LDL-induced cytotoxicity. Circ. Res. 1999, 84, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhou, Q.; Song, Y.; Wu, W.; Yu, H.; Wang, S.; Chen, Y.; Ye, M.; Lu, L. Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS ONE 2013, 8, e82379. [Google Scholar] [CrossRef] [PubMed]
- Loidl, A.; Claus, R.; Ingolic, E.; Deigner, H.-P.; Hermetter, A. Role of ceramide in activation of stress-associated MAP kinases by minimally modified LDL in vascular smooth muscle cells. Biochim. Biophys. Acta 2004, 1690, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.D.; Leitinger, N.; Navab, M.; Faull, K.F.; Hörkkö, S.; Witztum, J.L.; Palinski, W.; Schwenke, D.; Salomon, R.G.; Sha, W.; et al. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J. Biol. Chem. 1997, 272, 13597–13607. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I.; Choi, S.-H.; Wiesner, P.; Fang, L.; Harkewicz, R.; Hartvigsen, K.; Boullier, A.; Gonen, A.; Diehl, C.J.; Que, X.; et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 2011, 108, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Boullier, A.; Li, Y.; Quehenberger, O.; Palinski, W.; Tabas, I.; Witztum, J.L.; Miller, Y.I. Minimally oxidized LDL offsets the apoptotic effects of extensively oxidized LDL and free cholesterol in macrophages. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Vekshtein, V.; Gordon, H.M.; Tsuda, T. Angiotensin II-stimulated protein synthesis in cultured vascular smooth muscle cells. Hypertension 1989, 13, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Griendling, K.K. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J. Biol. Chem. 1998, 273, 15022–15029. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, M.; Yoshizumi, M.; Tsuchiya, K.; Kirima, K.; Tamaki, T. Antioxidants inhibit JNK and p38 MAPK activation but not ERK ½ activation by angiotensin II in rat aortic smooth muscle cells. Hypertens. Res. 2001, 24, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Golledge, J.; Heywood, E.B.; Bruemmer, D.; Daugherty, A. Regulation of peroxisome proliferator-activated receptor-γ by angiotensin II via transforming growth factor-β1-activated p38 mitogen-activated protein kinase in aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ha, C.-H.; Wong, C.; Wang, W.; Hausser, A.; Pfizenmaier, K.; Olson, E.N.; McKinsey, T.A.; Jin, Z.-G. Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Lee, C.-K.; Lee, S.H.; Roh, H.Y.; Bae, Y.M.; Lee, K.-Y.; Lim, J.; Park, P.-J.; Park, T.-K.; Lee, Y.L.; et al. p38 mitogen-activated protein kinase contributes to angiotensin II-stimulated migration of rat aortic smooth muscle cells. J. Pharmacol. Sci. 2007, 105, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Won, S.-M.; Park, Y.-H.; Kim, H.-J.; Park, K.-M.; Lee, W.-J. Catechins inhibit angiotensin II-induced vascular smooth muscle cell proliferation via mitogen-activated protein kinase pathway. Exp. Mol. Med. 2006, 38, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Bairati, A.; DeBiasi, S. Presence of a smooth muscle system in aortic valve leaflets. Anat. Embryol. 1981, 161, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Della Rocca, F.; Sartore, S.; Guidolin, D.; Bertiplaglia, B.; Gerosa, G.; Casarotto, D.; Pauletto, P. Cell composition of the human pulmonary valve: A comparative study with the aortic valve—The VESALIO Project. Ann. Thorac. Surg. 2000, 70, 1594–1600. [Google Scholar] [CrossRef]
- Latif, N.; Sarathchandra, P.; Chester, A.H.; Yacoub, M.H. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur. Heart J. 2015, 36, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Jian, B.; Narula, N.; Li, Q.-Y.; Mohler, E.R.; Levy, R.J. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann. Thorac. Surg. 2003, 75, 457–465. [Google Scholar] [CrossRef]
- Osman, L.; Yacoub, M.H.; Latif, N.; Amrani, M.; Chester, A.H. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 2006, 114, I547–I552. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Kusanagi, K.; Inoue, H. Divergence and convergence of TGF-beta/BMP signaling. J. Cell. Physiol. 2001, 187, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Holmes, A.; Murphy, G.A.; Mishra, K.; Rosenkranz, A.C.; Horowitz, J.D.; Kennedy, J.A. TGF-beta1-Induced MAPK activation promotes collagen synthesis, nodule formation, redox stress and cellular senescence in porcine aortic valve interstitial cells. J. Heart Valve Dis. 2013, 22, 621–630. [Google Scholar] [PubMed]
- Lee, K.-S.; Hong, S.-H.; Bae, S.-C. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene 2002, 21, 7156–7163. [Google Scholar] [CrossRef] [PubMed]
- Gallea, S.; Lallemand, F.; Atfi, A.; Rawadi, G.; Ramez, V.; Spinella-Jaegle, S.; Kawai, S.; Faucheu, C.; Huet, L.; Baron, R.; et al. Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone 2001, 28, 491–498. [Google Scholar] [CrossRef]
- Song, R.; Zeng, Q.; Ao, L.; Yu, J.A.; Cleveland, J.C.; Zhao, K.-S.; Fullerton, D.A.; Meng, X. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll-like receptor-2. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, Z.; Cai, Z.; Dong, N.; Liu, Y. Oxidized low-density lipoprotein promotes osteoblastic differentiation of valvular interstitial cells through RAGE/MAPK. Cardiology 2015, 130, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pisonero, I.; López, J.; Onecha, E.; Dueñas, A.I.; Maeso, P.; Crespo, M.S.; San Román, J.A.; García-Rodríguez, C. Synergy between sphingosine 1-phosphate and lipopolysaccharide signaling promotes an inflammatory, angiogenic and osteogenic response in human aortic valve interstitial cells. PLoS ONE 2014, 9, e109081. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Ji, J.; Li, L.; Chen, R.; Hu, W.-C. Adventitial fibroblasts are activated in the early stages of atherosclerosis in the apolipoprotein E knockout mouse. Biochem. Biophys. Res. Commun. 2007, 352, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Dobreva, I.; Waeber, G.; Mooser, V.; James, R.W.; Widmann, C. LDLs induce fibroblast spreading independently of the LDL receptor via activation of the p38 MAPK pathway. J. Lipid Res. 2003, 44, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Dobreva, I.; Zschörnig, O.; Waeber, G.; James, R.W.; Widmann, C. Cholesterol is the major component of native lipoproteins activating the p38 mitogen-activated protein kinases. Biol. Chem. 2005, 386, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Gerrity, R.G. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am. J. Pathol. 1981, 103, 181–190. [Google Scholar] [PubMed]
- Qin, X.; Corriere, M.A.; Matrisian, L.M.; Guzman, R.J. Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, J.-O.; Aikawa, E.; Libby, P.; Vachon, J.R.; Inada, M.; Krane, S.M.; Whittaker, P.; Aikawa, M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation 2005, 112, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Sosnovik, D.; Lok, V.M.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007, 115, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, Y.; Wang, X.; New, L.; Han, J.; Brunk, U.T. Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS 2002, 110, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Twardowski, L.; Fehr, S.; Aner, C.; Schaeffeler, E.; Joos, T.; Knorpp, T.; Dorweiler, B.; Laufer, S.; Schwab, M.; et al. Selective p38α MAP kinase/MAPK14 inhibition in enzymatically modified LDL-stimulated human monocytes: Implications for atherosclerosis. FASEB J. 2017, 31, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Shafi, S.; Codrington, R.; Gidden, L.M.; Ferns, G.A.A. Increased expression of phosphorylated forms of heat-shock protein-27 and p38MAPK in macrophage-rich regions of fibro-fatty atherosclerotic lesions in the rabbit. Int. J. Exp. Pathol. 2016, 97, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z. OxLDL upregulates CXCR2 expression in monocytes via scavenger receptors and activation of p38 mitogen-activated protein kinase. Cardiovasc. Res. 2002, 53, 524–532. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, H.; Feng, T.; Wu, R.; Sun, X.; Guan, N.; Qu, L.; Gao, Z.; Yan, J.; Xu, N.; et al. β-Glucan attenuates inflammatory responses in oxidized LDL-induced THP-1 cells via the p38 MAPK pathway. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Hakala, J.K.; Lindstedt, K.A.; Kovanen, P.T.; Pentikäinen, M.O. Low-Density Lipoprotein Modified by Macrophage-Derived Lysosomal Hydrolases Induces Expression and Secretion of IL-8 Via p38 MAPK and NF-κB by Human Monocyte-Derived Macrophages. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2504–2509. [Google Scholar] [CrossRef] [PubMed]
- Senokuchi, T.; Matsumura, T.; Sakai, M.; Matsuo, T.; Yano, M.; Kiritoshi, S.; Sonoda, K.; Kukidome, D.; Nishikawa, T.; Araki, E. Extracellular signal-regulated kinase and p38 mitogen-activated protein kinase mediate macrophage proliferation induced by oxidized low-density lipoprotein. Atherosclerosis 2004, 176, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Wang, Y.; Han, S.; Senokuchi, T.; Schrijvers, D.M.; Kuriakose, G.; Tall, A.R.; Tabas, I.A. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J. Clin. Investig. 2009, 119, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Jagavelu, K.; Tietge, U.J.F.; Gaestel, M.; Drexler, H.; Schieffer, B.; Bavendiek, U. Systemic deficiency of the MAP kinase-activated protein kinase 2 reduces atherosclerosis in hypercholesterolemic mice. Circ. Res. 2007, 101, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Moriya, J. Critical roles of inflammation in atherosclerosis. J. Cardiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Towler, D.A. Oxidation, inflammation, and aortic valve calcification peroxide paves an osteogenic path. J. Am. Coll. Cardiol. 2008, 52, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Helske, S.; Lindstedt, K.A.; Laine, M.; Mäyränpää, M.; Werkkala, K.; Lommi, J.; Turto, H.; Kupari, M.; Kovanen, P.T. Induction of local angiotensin II-producing systems in stenotic aortic valves. J. Am. Coll. Cardiol. 2004, 44, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation 1994, 90, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Yan, C.; Deng, Q.; Dong, X.; Duan, Z.-M.; Gao, D.-F.; Niu, X.-L. Oxidized low-density lipoprotein induces inflammatory responses in cultured human mast cells via Toll-like receptor 4. Cell. Physiol. Biochem. 2013, 31, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Dalsgaard, C.J.; Haegerstrand, A.; Rosenqvist, M.; Rydén, L.; Nilsson, J. Accumulation of T lymphocytes and expression of interleukin-2 receptors in nonrheumatic stenotic aortic valves. J. Am. Coll. Cardiol. 1994, 23, 1162–1170. [Google Scholar] [CrossRef]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 1986, 6, 131–138. [Google Scholar] [CrossRef]
- Mazzone, A.; Epistolato, M.C.; de Caterina, R.; Storti, S.; Vittorini, S.; Sbrana, S.; Gianetti, J.; Bevilacqua, S.; Glauber, M.; Biagini, A.; et al. Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J. Am. Coll. Cardiol. 2004, 43, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Lei, Y.; Martínez-Martínez, E.; Body, S.C.; Schlotter, F.; Creager, M.; Assmann, A.; Khabbaz, K.; Libby, P.; Hansson, G.K.; et al. Interferon-γ Released by Activated CD8+ T Lymphocytes Impairs the Calcium Resorption Potential of Osteoclasts in Calcified Human Aortic Valves. Am. J. Pathol. 2017, 187, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Salojin, K.V.; Gao, J.X.; Cameron, M.J.; Bergerot, I.; Delovitch, T.L. p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J. Immunol. 1999, 162, 3819–3829. [Google Scholar] [PubMed]
- Rincón, M.; Enslen, H.; Raingeaud, J.; Recht, M.; Zapton, T.; Su, M.S.; Penix, L.A.; Davis, R.J.; Flavell, R.A. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998, 17, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Melián, A.; Geng, Y.J.; Sukhova, G.K.; Libby, P.; Porcelli, S.A. CD1 expression in human atherosclerosis. A potential mechanism for T cell activation by foam cells. Am. J. Pathol. 1999, 155, 775–786. [Google Scholar] [CrossRef]
- Tupin, E.; Nicoletti, A.; Elhage, R.; Rudling, M.; Ljunggren, H.-G.; Hansson, G.K.; Berne, G.P. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J. Exp. Med. 2004, 199, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; To, K.; Kanellakis, P.; Hosseini, H.; Deswaerte, V.; Tipping, P.; Smyth, M.J.; Toh, B.-H.; Bobik, A.; Kyaw, T. CD4+ natural killer T cells potently augment aortic root atherosclerosis by perforin- and granzyme B-dependent cytotoxicity. Circ. Res. 2015, 116, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kyriakakis, E.; Cavallari, M.; Andert, J.; Philippova, M.; Koella, C.; Bochkov, V.; Erne, P.; Wilson, S.B.; Mori, L.; Biedermann, B.C.; et al. Invariant natural killer T cells: Linking inflammation and neovascularization in human atherosclerosis. Eur. J. Immunol. 2010, 40, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Kyriakakis, E.; Cavallari, M.; Pfaff, D.; Fabbro, D.; Mestan, J.; Philippova, M.; de Libero, G.; Erne, P.; Resink, T.J. IL-8-mediated angiogenic responses of endothelial cells to lipid antigen activation of iNKT cells depend on EGFR transactivation. J. Leukoc. Biol. 2011, 90, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Shuvy, M.; Ben Ya’acov, A.; Zolotarov, L.; Lotan, C.; Ilan, Y. Beta glycosphingolipids suppress rank expression and inhibit natural killer T cell and CD8+ accumulation in alleviating aortic valve calcification. Int. J. Immunopathol. Pharmacol. 2009, 22, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.; Mielimonka, A.; Natorska, J.; Wypasek, E.; Gawęda, B.; Sobczyk, D.; Kapusta, P.; Malinowski, K.P.; Kapelak, B. Lymphocyte and monocyte subpopulations in severe aortic stenosis at the time of surgical intervention. Cardiovasc. Pathol. 2018, 35, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bessoles, S.; Fouret, F.; Dudal, S.; Besra, G.S.; Sanchez, F.; Lafont, V. IL-2 triggers specific signaling pathways in human NKT cells leading to the production of pro- and anti-inflammatory cytokines. J. Leukoc. Biol. 2008, 84, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Nagaleekar, V.K.; Sabio, G.; Aktan, I.; Chant, A.; Howe, I.W.; Thornton, T.M.; Benoit, P.J.; Davis, R.J.; Rincon, M.; Boyson, J.E. Translational control of NKT cell cytokine production by p38 MAPK. J. Immunol. 2011, 186, 4140–4146. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.K.; Bisch, S.P.; Leon-Ponte, M.; Hayatsu, J.; Mazzuca, D.M.; Maleki Vareki, S.; Haeryfar, S.M.M. Negative modulation of invariant natural killer T cell responses to glycolipid antigens by p38 MAP kinase. Int. Immunopharmacol. 2010, 10, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Houtkamp, M.A.; de Boer, O.J.; van der Loos, C.M.; van der Wal, A.C.; Becker, A.E. Adventitial infiltrates associated with advanced atherosclerotic plaques: Structural organization suggests generation of local humoral immune responses. J. Pathol. 2001, 193, 263–269. [Google Scholar] [CrossRef]
- Ramshaw, A.L.; Parums, D.V. Immunohistochemical characterization of inflammatory cells associated with advanced atherosclerosis. Histopathology 1990, 17, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Wallby, L.; Steffensen, T.; Jonasson, L.; Broqvist, M. Inflammatory Characteristics of Stenotic Aortic Valves: A Comparison between Rheumatic and Nonrheumatic Aortic Stenosis. Cardiol. Res. Pract. 2013, 2013, 895215. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Hörkkö, S.; Chang, M.K.; Curtiss, L.K.; Palinski, W.; Silverman, G.J.; Witztum, J.L. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Investig. 2000, 105, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Hörkkö, S.; Miller, E.; Steinbrecher, U.P.; Powell, H.C.; Curtiss, L.K.; Witztum, J.L. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Investig. 1996, 98, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Shaw, P.X.; Chang, M.-K.; Boullier, A.; Hartvigsen, K.; Hörkkö, S.; Miller, Y.I.; Woelkers, D.A.; Corr, M.; Witztum, J.L. The role of natural antibodies in atherogenesis. J. Lipid Res. 2005, 46, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Hörkkö, S.; Dewan, A.; Chang, M.-K.; Kieu, E.P.; Goodyear, C.S.; Shaw, P.X.; Palinski, W.; Witztum, J.L.; Silverman, G.J. Pneumococcal vaccination decreases atherosclerotic lesion formation: Molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 2003, 9, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.-Y.; Fogelstrand, L.; Hartvigsen, K.; Hansen, L.F.; Woelkers, D.; Shaw, P.X.; Choi, J.; Perkmann, T.; Bäckhed, F.; Miller, Y.I.; et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Investig. 2009, 119, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Tay, C.; Khan, A.; Dumouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Diehl, C.J.; Witztum, J.L.; Binder, C.J. B cells and humoral immunity in atherosclerosis. Circ. Res. 2014, 114, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Khiem, D.; Cyster, J.G.; Schwarz, J.J.; Black, B.L. A p38 MAPK-MEF2C pathway regulates B-cell proliferation. Proc. Natl. Acad. Sci. USA 2008, 105, 17067–17072. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.-S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 2009, 206, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Lord, R.S. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc. Res. 1995, 29, 689–696. [Google Scholar] [CrossRef]
- Yilmaz, A.; Lochno, M.; Traeg, F.; Cicha, I.; Reiss, C.; Stumpf, C.; Raaz, D.; Anger, T.; Amann, K.; Probst, T.; et al. Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques. Atherosclerosis 2004, 176, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, E.K.; Ley, K. How dendritic cells shape atherosclerosis. Trends Immunol. 2011, 32, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Arrighi, J.F.; Rebsamen, M.; Rousset, F.; Kindler, V.; Hauser, C. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha, and contact sensitizers. J. Immunol. 2001, 166, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, M.; Wang, S.; Hong, B.; Wang, Z.; Li, H.; Zheng, Y.; Yang, J.; Davis, R.E.; Qian, J.; et al. p38 MAPK-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat. Commun. 2014, 5, 4229. [Google Scholar] [CrossRef] [PubMed]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Rossebø, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Bärwolf, C.; Holme, I.; Kesäniemi, Y.A.; et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Tall, A.R.; Daemen, M.J.A.P.; Falk, E.; Fisher, E.A.; García-Cardeña, G.; Lusis, A.J.; Owens, A.P.; Rosenfeld, M.E.; Virmani, R. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Circ. Res. 2017, 121, e53–e79. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Romir, J.; Fischer, S.; Koeberle, A.; Schattel, V.; Albrecht, W.; Grütter, C.; Werz, O.; Rauh, D.; Stehle, T.; et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2012, 8, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Sarov-Blat, L.; Morgan, J.M.; Fernandez, P.; James, R.; Fang, Z.; Hurle, M.R.; Baidoo, C.; Willette, R.N.; Lepore, J.J.; Jensen, S.E.; et al. Inhibition of p38 mitogen-activated protein kinase reduces inflammation after coronary vascular injury in humans. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Elkhawad, M.; Rudd, J.H.F.; Sarov-Blat, L.; Cai, G.; Wells, R.; Davies, L.C.; Collier, D.J.; Marber, M.S.; Choudhury, R.P.; Fayad, Z.A.; et al. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. JACC Cardiovasc. Imaging 2012, 5, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Newby, L.K.; Marber, M.S.; Melloni, C.; Sarov-Blat, L.; Aberle, L.H.; Aylward, P.E.; Cai, G.; de Winter, R.J.; Hamm, C.W.; Heitner, J.F.; et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: A randomised phase 2 trial. Lancet 2014, 384, 1187–1195. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.A.; Gutierrez, J.A.T.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2016, 315, 1591–1599. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. https://doi.org/10.3390/ijms19123761
Reustle A, Torzewski M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. International Journal of Molecular Sciences. 2018; 19(12):3761. https://doi.org/10.3390/ijms19123761
Chicago/Turabian StyleReustle, Anna, and Michael Torzewski. 2018. "Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis" International Journal of Molecular Sciences 19, no. 12: 3761. https://doi.org/10.3390/ijms19123761
APA StyleReustle, A., & Torzewski, M. (2018). Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. International Journal of Molecular Sciences, 19(12), 3761. https://doi.org/10.3390/ijms19123761