Translation Stress Regulates Ribosome Synthesis and Cell Proliferation



{kind=link}
{kind=link}
Abstract
1. Human Ribosome Biogenesis—Overview
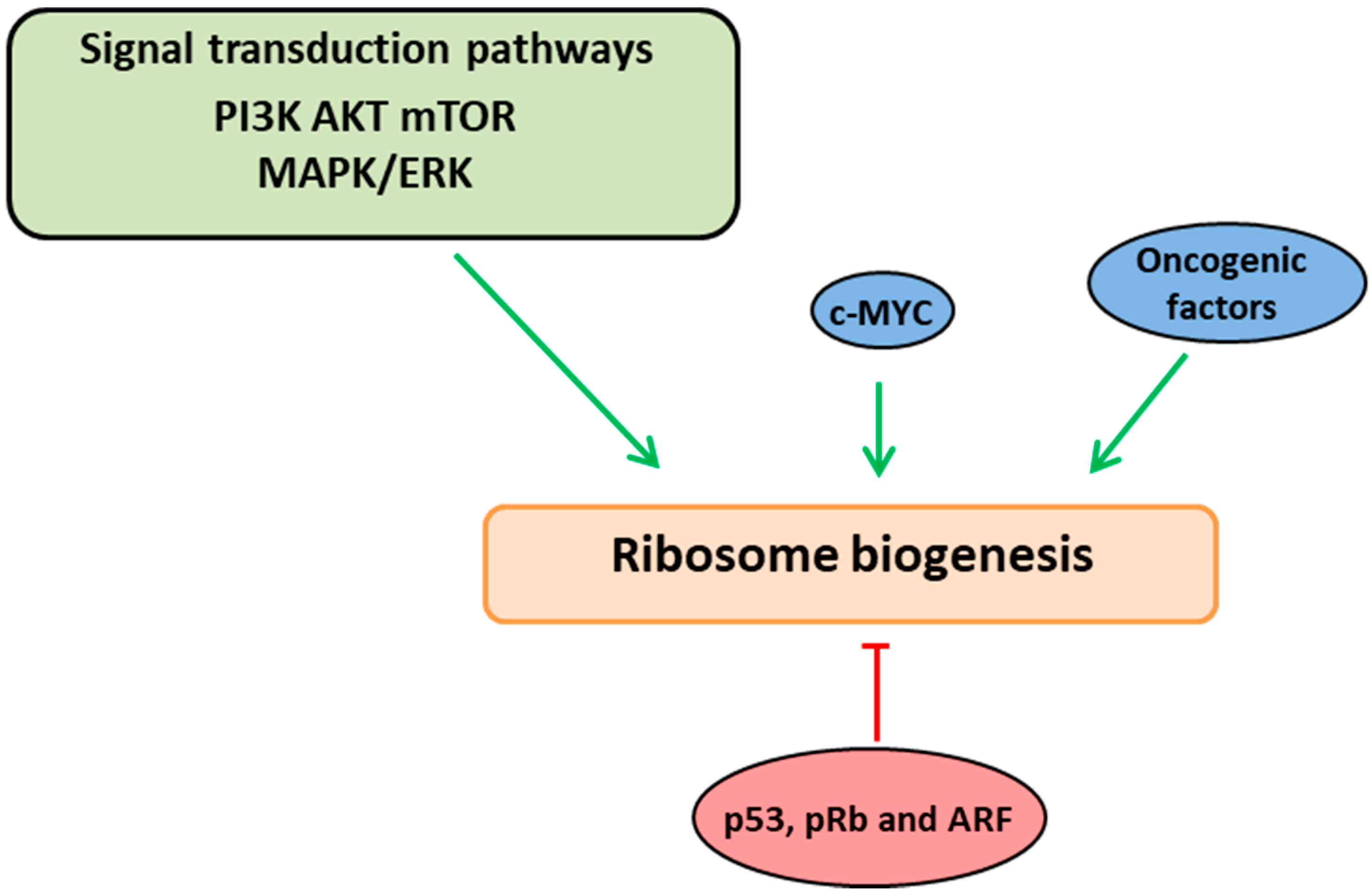
2. Ribosome Biogenesis Nexus with Cell Proliferation
3. Ribosomopathies and Cancer
4. mRNA Translation Stress and Feedback to Ribosome Biogenesis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schmeing, T.M.; Ramakrishnan, V. What recent ribosome structures have revealed about the mechanism of translation. Nature 2009, 461, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Armache, J.P.; Jarasch, A.; Anger, A.M.; Villa, E.; Becker, T.; Bhushan, S.; Jossinet, F.; Habeck, M.; Dindar, G.; Franckenberg, S.; et al. Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-A resolution. Proc. Natl. Acad. Sci. USA 2010, 107, 19748–19753. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, S.; Ben-Shem, A.; Garreau de Loubresse, N.; Jenner, L.; Yusupova, G.; Yusupov, M. One core, two shells: Bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Rabl, J.; Leibundgut, M.; Ataide, S.F.; Haag, A.; Ban, N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 2011, 331, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Klinge, S.; Voigts-Hoffmann, F.; Leibundgut, M.; Arpagaus, S.; Ban, N. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science 2011, 334, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Yusupova, G.; Yusupov, M. High-resolution structure of the eukaryotic 80S ribosome. Annu. Rev. Biochem. 2014, 83, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, P.; Topf, M.; Menetret, J.F.; Eswar, N.; Cannone, J.J.; Gutell, R.R.; Sali, A.; Akey, C.W. Structure of the mammalian 80S ribosome at 8.7 A resolution. Structure 2008, 16, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Woolford, J.L., Jr.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef] [PubMed]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Natchiar, S.K.; Myasnikov, A.G.; Hazemann, I.; Klaholz, B.P. Visualizing the Role of 2’-OH rRNA Methylations in the Human Ribosome Structure. Biomolecules 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim. Biophys. Acta 2015, 1849, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Tschochner, H.; Hurt, E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 2003, 13, 255–263. [Google Scholar] [CrossRef]
- Ogle, J.M.; Ramakrishnan, V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005, 74, 129–177. [Google Scholar] [CrossRef] [PubMed]
- Zaher, H.S.; Green, R. Fidelity at the molecular level: Lessons from protein synthesis. Cell 2009, 136, 746–762. [Google Scholar] [CrossRef] [PubMed]
- Tafforeau, L.; Zorbas, C.; Langhendries, J.L.; Mullineux, S.T.; Stamatopoulou, V.; Mullier, R.; Wacheul, L.; Lafontaine, D.L. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol. Cell 2013, 51, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Wild, T.; Horvath, P.; Wyler, E.; Widmann, B.; Badertscher, L.; Zemp, I.; Kozak, K.; Csucs, G.; Lund, E.; Kutay, U. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010, 8, e1000522. [Google Scholar] [CrossRef] [PubMed]
- Badertscher, L.; Wild, T.; Montellese, C.; Alexander, L.T.; Bammert, L.; Sarazova, M.; Stebler, M.; Csucs, G.; Mayer, T.U.; Zamboni, N.; et al. Genome-wide RNAi Screening Identifies Protein Modules Required for 40S Subunit Synthesis in Human Cells. Cell Rep. 2015, 13, 2879–2891. [Google Scholar] [CrossRef] [PubMed]
- O’Donohue, M.F.; Choesmel, V.; Faubladier, M.; Fichant, G.; Gleizes, P.E. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J. Cell Biol. 2010, 190, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Farley, K.I.; Baserga, S.J. Probing the mechanisms underlying human diseases in making ribosomes. Biochem. Soc. Trans. 2016, 44, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Bodem, J.; Dobreva, G.; Hoffmann-Rohrer, U.; Iben, S.; Zentgraf, H.; Delius, H.; Vingron, M.; Grummt, I. TIF-IA, the factor mediating growth-dependent control of ribosomal RNA synthesis, is the mammalian homolog of yeast Rrn3p. EMBO Rep. 2000, 1, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Trere, D. Ribosome biogenesis and cancer. Acta Histochem. 2017, 119, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Brighenti, E.; Trere, D.; Derenzini, M. Targeted cancer therapy with ribosome biogenesis inhibitors: A real possibility? Oncotarget 2015, 6, 38617–38627. [Google Scholar] [CrossRef] [PubMed]
- Stepanchick, A.; Zhi, H.; Cavanaugh, A.H.; Rothblum, K.; Schneider, D.A.; Rothblum, L.I. DNA binding by the ribosomal DNA transcription factor rrn3 is essential for ribosomal DNA transcription. J. Biol. Chem. 2013, 288, 9135–9144. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Hurt, E.; Bassler, J. Driving ribosome assembly. Biochim. Biophys. Acta 2010, 1803, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. Wisely chosen paths–regulation of rRNA synthesis: Delivered on 30 June 2010 at the 35th FEBS Congress in Gothenburg, Sweden. FEBS J. 2010, 277, 4626–4639. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, J.; Karbstein, K.; Woolford, J.L., Jr. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu. Rev. Biochem. 2015, 84, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G. An encore for ribosome biogenesis in the control of cell proliferation. Nat. Cell Biol. 2000, 2, E71–E72. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, S.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Thomas, G.; Mercer, C.A. Growth control and ribosomopathies. Curr. Opin. Genet. Dev. 2013, 23, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Stefanovsky, V.Y.; Pelletier, G.; Hannan, R.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell 2001, 8, 1063–1073. [Google Scholar] [CrossRef]
- Stefanovsky, V.; Langlois, F.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol. Cell 2006, 21, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Plas, D.R.; Thomas, G. Tubers and tumors: Rapamycin therapy for benign and malignant tumors. Curr. Opin. Cell Biol. 2009, 21, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell. Biol. 2003, 23, 8862–8877. [Google Scholar] [CrossRef] [PubMed]
- Kantidakis, T.; Ramsbottom, B.A.; Birch, J.L.; Dowding, S.N.; White, R.J. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc. Natl. Acad. Sci. USA 2010, 107, 11823–11828. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: MTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed]
- Iadevaia, V.; Liu, R.; Proud, C.G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Sem. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, N.; Felton-Edkins, Z.A.; Kenneth, N.S.; Goodfellow, S.J.; Athineos, D.; Zhang, J.; Ramsbottom, B.A.; Innes, F.; Kantidakis, T.; Kerr, E.R.; et al. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem. Soc. Symp. 2006, 73, 141–154. [Google Scholar] [CrossRef]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Sirri, V.; Roussel, P.; Hernandez-Verdun, D. In vivo release of mitotic silencing of ribosomal gene transcription does not give rise to precursor ribosomal RNA processing. J. Cell Biol. 2000, 148, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Heix, J.; Vente, A.; Voit, R.; Budde, A.; Michaelidis, T.M.; Grummt, I. Mitotic silencing of human rRNA synthesis: Inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO J. 1998, 17, 7373–7381. [Google Scholar] [CrossRef] [PubMed]
- Sirri, V.; Roussel, P.; Hernandez-Verdun, D. The mitotically phosphorylated form of the transcription termination factor TTF-1 is associated with the repressed rDNA transcription machinery. J. Cell Sci. 1999, 112 Pt 19, 3259–3268. [Google Scholar]
- Voit, R.; Seiler, J.; Grummt, I. Cooperative Action of Cdk1/cyclin B and SIRT1 Is Required for Mitotic Repression of rRNA Synthesis. PLoS Genet. 2015, 11, e1005246. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, A.H.; Hempel, W.M.; Taylor, L.J.; Rogalsky, V.; Todorov, G.; Rothblum, L.I. Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 1995, 374, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.; Schafer, K.; Grummt, I. Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol. Cell. Biol. 1997, 17, 4230–4237. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Hannan, R.D.; Smith, S.D.; Jefferson, L.S.; Lun, M.; Rothblum, L.I. Rb and p130 regulate RNA polymerase I transcription: Rb disrupts the interaction between UBF and SL-1. Oncogene 2000, 19, 4988–4999. [Google Scholar] [CrossRef] [PubMed]
- Ciarmatori, S.; Scott, P.H.; Sutcliffe, J.E.; McLees, A.; Alzuherri, H.M.; Dannenberg, J.H.; te Riele, H.; Grummt, I.; Voit, R.; White, R.J. Overlapping functions of the pRb family in the regulation of rRNA synthesis. Mol. Cell. Biol. 2001, 21, 5806–5814. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Trouche, D.; Martin, K.; Jackson, S.P.; Kouzarides, T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 1996, 382, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Felton-Edkins, Z.A.; Kenneth, N.S.; Brown, T.R.; Daly, N.L.; Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle 2003, 2, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Gnanasundram, S.V.; Pyndiah, S.; Daskalogianni, C.; Armfield, K.; Nylander, K.; Wilson, J.B.; Fahraeus, R. PI3Kδ activates E2F1 synthesis in response to mRNA translation stress. Nat. Commun. 2017, 8, 2103. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Whyte, P.; Buchkovich, K.J.; Horowitz, J.M.; Friend, S.H.; Raybuck, M.; Weinberg, R.A.; Harlow, E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988, 334, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Phelps, W.C.; Yee, C.L.; Munger, K.; Howley, P.M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell 1988, 53, 539–547. [Google Scholar] [CrossRef]
- Felsani, A.; Mileo, A.M.; Paggi, M.G. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 2006, 25, 5277–5285. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- David-Pfeuty, T. The flexible evolutionary anchorage-dependent Pardee’s restriction point of mammalian cells: How its deregulation may lead to cancer. Biochim. Biophys. Acta 2006, 1765, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Peddigari, S.; Mercer, C.A.; Thomas, G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013, 4, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 2014, 1842, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Zhang, Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 2010, 29, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Zambetti, G.P. Translational research? Ribosome integrity and a new p53 tumor suppressor checkpoint. Cell Death Differ. 2006, 13, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Cellular stress and nucleolar function. Cell Cycle 2005, 4, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q− syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Vlachos, A.; Atsidaftos, E.; Carlson-Donohoe, H.; Markello, T.C.; Arceci, R.J.; Ellis, S.R.; Lipton, J.M.; Bodine, D.M. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood 2011, 118, 6943–6951. [Google Scholar] [CrossRef] [PubMed]
- Shwachman, H.; Diamond, L.K.; Oski, F.A.; Khaw, K.T. The Syndrome of Pancreatic Insufficiency and Bone Marrow Dysfunction. J. Pediatr. 1964, 65, 645–663. [Google Scholar] [CrossRef]
- Bolze, A.; Mahlaoui, N.; Byun, M.; Turner, B.; Trede, N.; Ellis, S.R.; Abhyankar, A.; Itan, Y.; Patin, E.; Brebner, S.; et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science 2013, 340, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Gazda, H.T. Ribosomopathies: How a common root can cause a tree of pathologies. Dis. Models Mech. 2015, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Tahmasebi, S.; Khoutorsky, A.; Mathews, M.B.; Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Barna, M. Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012, 13, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. Heterogeneity and specialized functions of translation machinery: From genes to organisms. Nat. Rev. Genet. 2018, 19, 431–452. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Rost, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol. Cell 2017, 67, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Dosil, M. Ribosome biogenesis and cancer: Basic and translational challenges. Curr. Opin. Genet. Dev. 2018, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Hoogeveen, A.T.; Esposito, J.; Sacchi, N. Loss of MTG16a (CBFA2T3), a novel rDNA repressor, leads to increased ribogenesis and disruption of breast acinar morphogenesis. J. Cell. Mol. Med. 2010, 14, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Brighenti, E.; Calabrese, C.; Liguori, G.; Giannone, F.A.; Trere, D.; Montanaro, L.; Derenzini, M. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: A new pathway connecting inflammation to cancer. Oncogene 2014, 33, 4396–4406. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Bertoni, S.; Brighenti, E.; Vici, M.; Trere, D.; Volarevic, S.; Montanaro, L.; Derenzini, M. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene 2011, 30, 3274–3288. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforets, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.P.; Solano-Gonzalez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 2013, 24, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Catez, F.; Diaz, J.J. Ribosome heterogeneity in tumorigenesis: The rRNA point of view. Mol. Cell. Oncol. 2015, 2, e983755. [Google Scholar] [CrossRef] [PubMed]
- Monaco, P.L.; Marcel, V.; Diaz, J.J.; Catez, F. 2’-O-Methylation of Ribosomal RNA: Towards an Epitranscriptomic Control of Translation? Biomolecules 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Belin, S.; Beghin, A.; Solano-Gonzalez, E.; Bezin, L.; Brunet-Manquat, S.; Textoris, J.; Prats, A.C.; Mertani, H.C.; Dumontet, C.; Diaz, J.J. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS ONE 2009, 4, e7147. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, L.; Pacilli, A.; Sethi, R.; Penzo, M.; Schneider, R.J.; Trere, D.; Brigotti, M.; Montanaro, L. Dyskerin depletion increases VEGF mRNA internal ribosome entry site-mediated translation. Nucleic Acids Res. 2013, 41, 8308–8318. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.; Peng, G.; Brandenburger, Y.; Zollo, O.; Xu, W.; Rego, E.; Ruggero, D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006, 312, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Montanaro, L. Turning Uridines around: Role of rRNA Pseudouridylation in Ribosome Biogenesis and Ribosomal Function. Biomolecules 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, C.J.; Green, R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, E1392–E1398. [Google Scholar] [CrossRef] [PubMed]
- Pisarev, A.V.; Skabkin, M.A.; Pisareva, V.P.; Skabkina, O.V.; Rakotondrafara, A.M.; Hentze, M.W.; Hellen, C.U.; Pestova, T.V. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell 2010, 37, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Ding, Y.; Niemczyk, M.; Kudla, G.; Plotkin, J.B. Rate-limiting steps in yeast protein translation. Cell 2013, 153, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Tenson, T.; Ehrenberg, M. Regulatory nascent peptides in the ribosomal tunnel. Cell 2002, 108, 591–594. [Google Scholar] [CrossRef]
- Wen, J.D.; Lancaster, L.; Hodges, C.; Zeri, A.C.; Yoshimura, S.H.; Noller, H.F.; Bustamante, C.; Tinoco, I. Following translation by single ribosomes one codon at a time. Nature 2008, 452, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Simms, C.L.; Hudson, B.H.; Mosior, J.W.; Rangwala, A.S.; Zaher, H.S. An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep. 2014, 9, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Guydosh, N.R.; Green, R. Dom34 rescues ribosomes in 3′ untranslated regions. Cell 2014, 156, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Nagy, G.; Dotu, I.; Zhou, H.; Yang, X.L.; Schimmel, P.; Senju, S.; Nishimura, Y.; Chuang, J.H.; Ackerman, S.L. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 2014, 345, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Manoury, B.; Fahraeus, R. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 2003, 301, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Apcher, S.; Daskalogianni, C.; Manoury, B.; Fahraeus, R. Epstein Barr virus-encoded EBNA1 interference with MHC class I antigen presentation reveals a close correlation between mRNA translation initiation and antigen presentation. PLoS Pathog. 2010, 6, e1001151. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Zhong, J.; Lekieffre, L.; Cowieson, N.P.; Clancy, J.L.; Preiss, T.; Balasubramanian, S.; Khanna, R.; Tellam, J. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014, 10, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, C.; Sturge, J.; Bennett, D.C.; O’Hare, M.J.; Allen, W.E.; Bain, J.; Jones, G.E.; Vanhaesebroeck, B. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110δ. Cancer Res. 2003, 63, 1667–1675. [Google Scholar] [PubMed]
- Koyasu, S. The role of PI3K in immune cells. Nat. Immunol. 2003, 4, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Bilancio, A.; Thomas, M.; Pearce, W.; Gilfillan, A.M.; Tkaczyk, C.; Kuehn, N.; Gray, A.; Giddings, J.; Peskett, E.; et al. Essential role for the p110δ phosphoinositide 3-kinase in the allergic response. Nature 2004, 431, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.L.; Chandra, A.; Nejentsev, S.; Condliffe, A.M.; Okkenhaug, K. PI3Kδ and primary immunodeficiencies. Nat. Rev. Immunol. 2016, 16, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, E.; Rossi, A.; Trere, D. Treating hematological malignancies with drugs inhibiting ribosome biogenesis: When and why. J. Hematol. Oncol. 2018, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Kochert, K.; Zhang, B.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Lu, H.; Yasui, T.; Sharpe, A.; Warren, H.; Cahir-McFarland, E.; Bronson, R.; Hung, S.C.; Kieff, E. Epstein-Barr virus nuclear antigen 1 does not induce lymphoma in transgenic FVB mice. Proc. Natl. Acad. Sci. USA 2005, 102, 820–825. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadivel Gnanasundram, S.; Fåhraeus, R. Translation Stress Regulates Ribosome Synthesis and Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 3757. https://doi.org/10.3390/ijms19123757
Vadivel Gnanasundram S, Fåhraeus R. Translation Stress Regulates Ribosome Synthesis and Cell Proliferation. International Journal of Molecular Sciences. 2018; 19(12):3757. https://doi.org/10.3390/ijms19123757
Chicago/Turabian StyleVadivel Gnanasundram, Sivakumar, and Robin Fåhraeus. 2018. "Translation Stress Regulates Ribosome Synthesis and Cell Proliferation" International Journal of Molecular Sciences 19, no. 12: 3757. https://doi.org/10.3390/ijms19123757
APA StyleVadivel Gnanasundram, S., & Fåhraeus, R. (2018). Translation Stress Regulates Ribosome Synthesis and Cell Proliferation. International Journal of Molecular Sciences, 19(12), 3757. https://doi.org/10.3390/ijms19123757